Structural Characterization of TRAF6 N-terminal for Therapeutic Uses
, , , ,, , , , ,
BioRxiv 2023
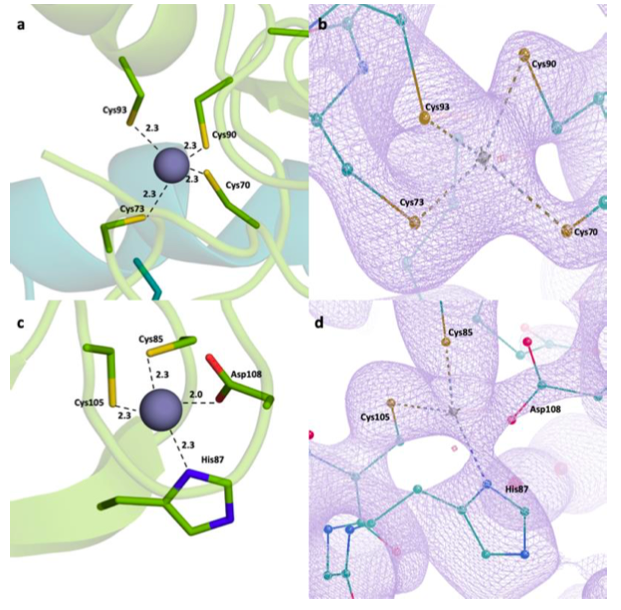
, , , ,, , , , ,
BioRxiv 2023
Tumor Necrosis Factor Receptor Associated Factors (TRAFs) are a protein family with a wide variety of roles and binding partners. Among them, TRAF6, a ubiquitin ligase, possesses unique receptor binding specificity and shows diverse functions in immune system regulation, cellular signaling, central nervous system (CNS), and tumor formation. TRAF6 consists of an N-terminal Really Interesting New Gene (RING) domain, multiple zinc fingers, and a C-terminal TRAF domain. RING domain and zinc fingers mediate the activation of nuclear factor kappa B (NF-κB), which has essentialroles in the regulation of inflammatory responses, proliferation, differentiation, migration, cell adhesion, and apoptosis. Therefore, it has been found that TRAF6 is overexpressed in various types of cancer including pancreatic, liver, lung, head and neck, breast, colorectal cancers, and melanoma along with inflammatory, autoimmune and neurodegenerative disorders. Furthermore, TRAF6 is an important therapeutic target for numerous disorders and structural studies of this protein are crucial for the development of next-generation therapeutics. Here, we present a TRAF6 N-terminal structure determined at the Turkish Light Source “Turkish DeLight” to 2.6 Å resolution at cryogenic temperature. This structure offers insight into the domain organization and zinc-binding, which are critical for protein function. Since the RING domain and the zinc fingers are key targets for TRAF6 therapeutics, structural insights are crucial for future research.
![]()
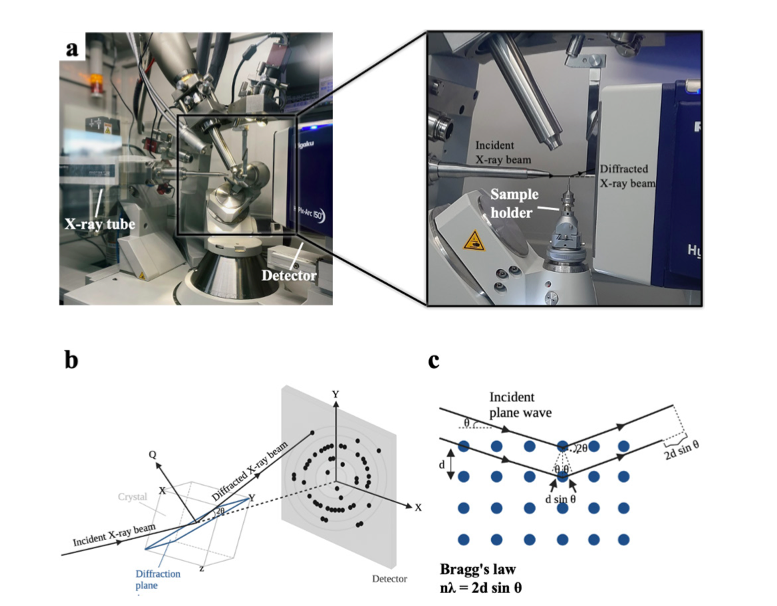
Atalay N, Akcan E, Gul M, Ayan E, Destan E, Ertem FB, Tokay N, Çakılkaya B, Nergiz N, Karakadioglu G, Kepceoglu A, Yapici I, Tosun B, Baldir N, Yildirim G, Johnson J, Guven O, Shafiei A, Arslan N, Yılmaz M, Kulakman C, Paydos S, Final S, Sabanoglu K, Pazarceviren A, Yilmaz A, Canbay B, Asci B, Kartal E, Tavli S, Caliseki M, Goc G, Mermer A, Yeşilay G, Altuntaş S, Tateishi H, Otsuka M, Fujita M, Tekin S, Ciftci H, Durdagi S, Dinler-Doganay G, Karaca Ezgi, Kaplan-Turkuoz B, Kabasakal B, Katı A, Demirci H
Turkish Journal of Biology 2023
X-ray crystallography is a robust and powerful structural biology technique that provides high-resolution atomic structures of biomacromolecules. Scientists use this technique to unravel mechanistic and structural details of biological macromolecules (e.g., proteins, nucleic acids, protein complexes, protein-nucleic acid complexes, or large biological compartments). Since its inception, single-crystal cryocrystallography has never been performed in Türkiye due to the lack of a single-crystal X-ray diffractometer. The X-ray diffraction facility recently established at the University of Health Sciences, İstanbul, Türkiye will enable Turkish and international researchers to easily perform high-resolution structural analysis of biomacromolecules from single crystals. Here, we describe the technical and practical outlook of a state-of-the-art home-source X-ray, using lysozyme as a model protein. The methods and practices described in this article can be applied to any biological sample for structural studies. Therefore, this article will be a valuable practical guide from sample preparation to data analysis.
![]()

B. Cakilkaya, I.H. Kavakli, H. DeMirci
JCB 2022
Photolyases (PLs) are the enzymes that reverse UV-Induced DNA damages by using blue-light as an energy source. Among those photolyases (6-4) PLs are the enzymes that repair (6-4) lesioned photoproduct. Here, we determined Vibrio cholerae (Vc) crystal structure of (6-4) PL at 2.5 Å resolution. Our high-resolution structure revealed that the presence of two well-known cofactors, flavin adenine dinucleotide (FAD) and or 6,7-dimethyl 8-ribityl-lumazin (DMRL), stably interact with an -helical and an / and domains, respectively. Additionally, it has also a third cofactor with distinct electron clouds corresponding to [4Fe-4S] cluster. Asp106 makes a hydrogen bond with the water and DMRL, which indicates further stabilization of the photoantenna DMRL to Vc(6-4) PL. Further analysis of Vc(6-4) PL structure revealed a possible region responsible for the DNA binding. The region located between residues 478-484 may binds the lesioned DNA and Arg483 forms a salt bridge with DNA to stabilize further Vc(6-4) PL with its substrate. Our comparative analysis revealed that DNA lesion could not bind to the Vc(6-4) PL in a similar fashion to the Dm(6-4) PL without a significant conformational change in the protein. The 23rd helix of the bacterial (6-4) PLs seems to have a remarkable plasticity and conformational changes facilitate the DNA binding.
![]()
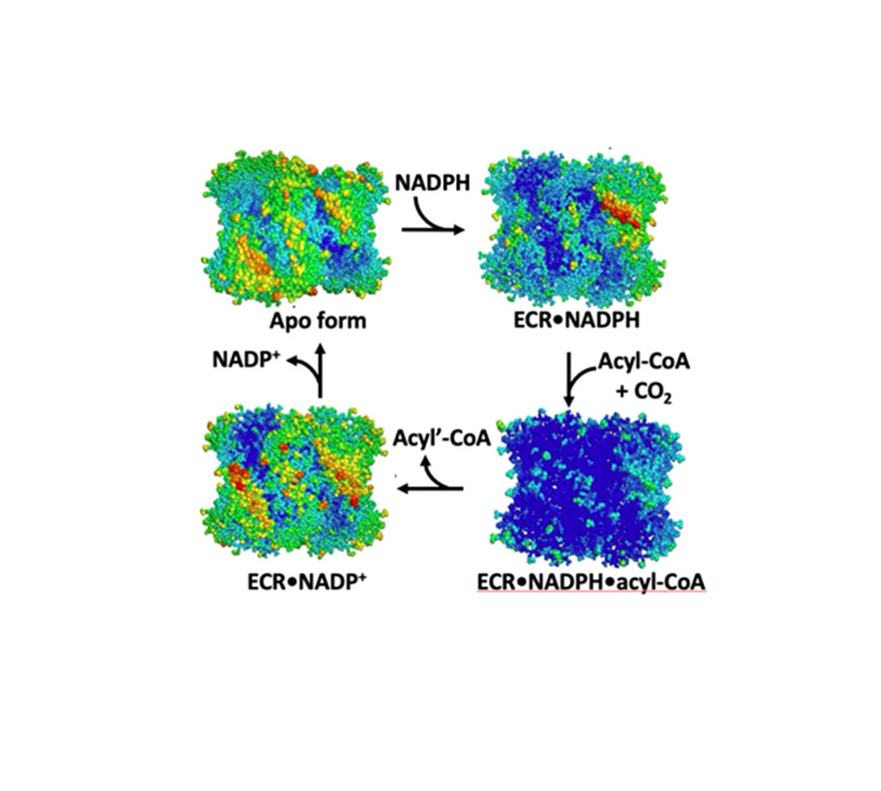
H. DeMirci, Y. Rao, G. M. Stoffel, B. Vögeli, K. Schell, A. Gomez, A. Batyuk, C. Gati, R.G. Sierra, M.S. Hunter, E. H. Dao, H. I. Ciftci, B. Hayes, F. Poitevin, P. Li, M. Kaur, K. Tono, D. Adrian Saez, S. Deutsch, Y. Yoshikuni, H. Grubmüller, T. J. Erb, E. Vöhringer-Martinez, and S. Wakatsuki
ACS Central Science 2022
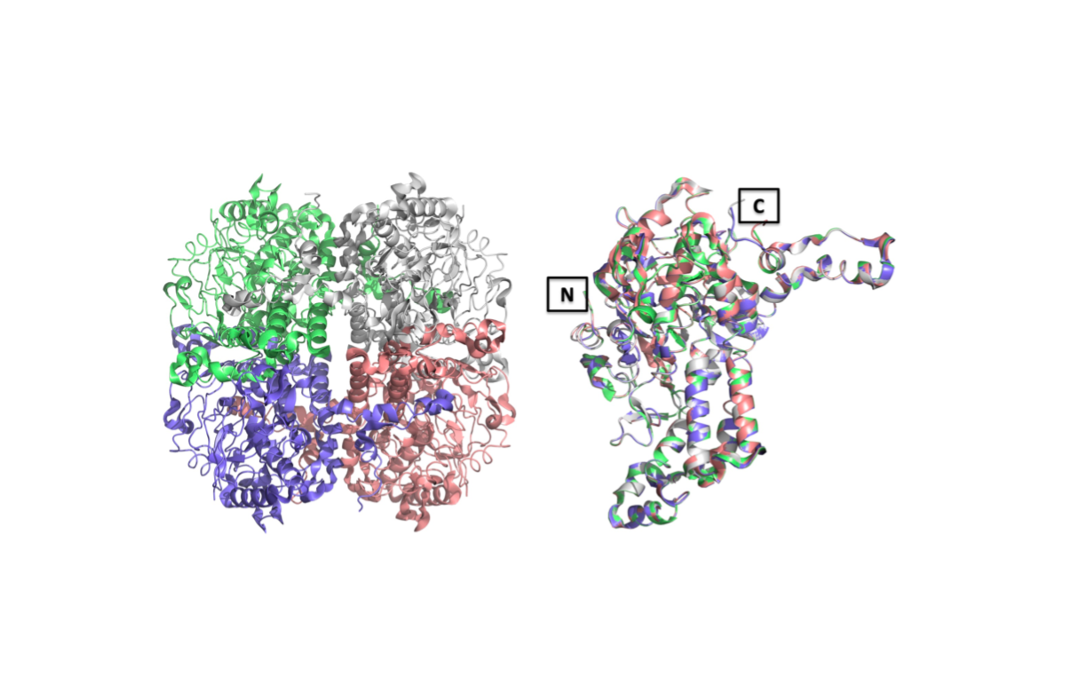
Enoyl-CoA carboxylases/reductases (ECRs) are some of the most efficient CO2-fixing enzymes described to date. However, the molecular mechanisms underlying the extraordinary catalytic activity of ECRs on the level of the protein assembly remain elusive. Here we used a combination of ambient-temperature X-ray free electron laser (XFEL) and cryogenic synchrotron experiments to study the structural organization of the ECR from Kitasatospora setae. The K. setae ECR is a homotetramer that differentiates into a pair of dimers of open- and closed-form subunits in the catalytically active state. Using molecular dynamics simulations and structure-based mutagenesis, we show that catalysis is synchronized in the K. setae ECR across the pair of dimers. This conformational coupling of catalytic domains is conferred by individual amino acids to achieve high CO2-fixation rates. Our results provide unprecedented insights into the dynamic organization and synchronized inter- and intrasubunit communications of this remarkably efficient CO2-fixing enzyme during catalysis.
![]()
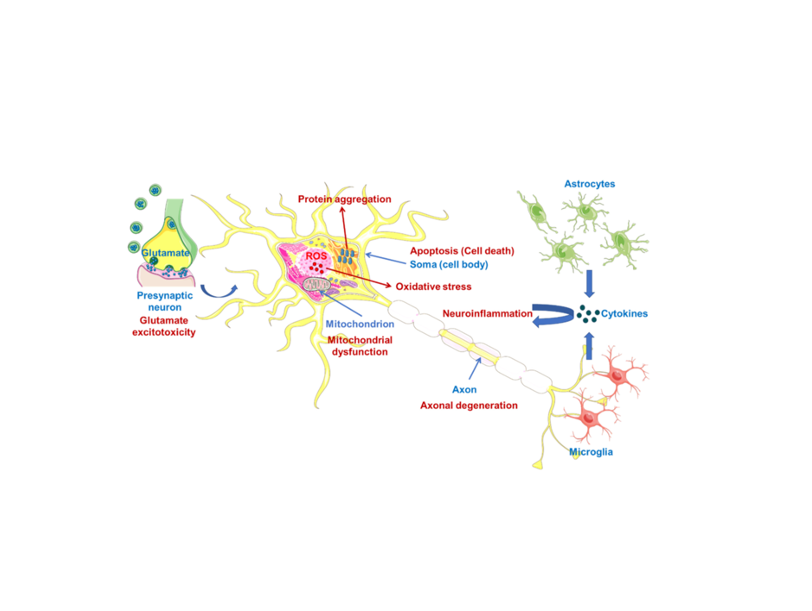
Sever B., Ciftci H., DeMirci H., Sever H., Ocak F., Yulug B., Tateishi H., Tateishi T., Otsuka M., Fujita M. and Başak A.N.
International Journal of Molecular Sciences 2022
Amyotrophic lateral sclerosis (ALS) is a rapidly debilitating fatal neurodegenerative disor- der, causing muscle atrophy and weakness, which leads to paralysis and eventual death. ALS has a multifaceted nature affected by many pathological mechanisms, including oxidative stress (also via protein aggregation), mitochondrial dysfunction, glutamate-induced excitotoxicity, apoptosis, neuroinflammation, axonal degeneration, skeletal muscle deterioration and viruses. This complexity is a major obstacle in defeating ALS. At present, riluzole and edaravone are the only drugs that have passed clinical trials for the treatment of ALS, notwithstanding that they showed modest benefits in a limited population of ALS. A dextromethorphan hydrobromide and quinidine sulfate combination was also approved to treat pseudobulbar affect (PBA) in the course of ALS. Globally, there is a struggle to prevent or alleviate the symptoms of this neurodegenerative disease, including implementation of antisense oligonucleotides (ASOs), induced pluripotent stem cells (iPSCs), CRISPR-9/Cas technique, non-invasive brain stimulation (NIBS) or ALS-on-a-chip technology. Additionally, researchers have synthesized and screened new compounds to be effective in ALS beyond the drug repurposing strategy. Despite all these efforts, ALS treatment is largely limited to palliative care, and there is a strong need for new therapeutics to be developed. This review focuses on and discusses which therapeutic strategies have been followed so far and what can be done in the future for the treatment of ALS.
![]()
,
Star Protocols 2022
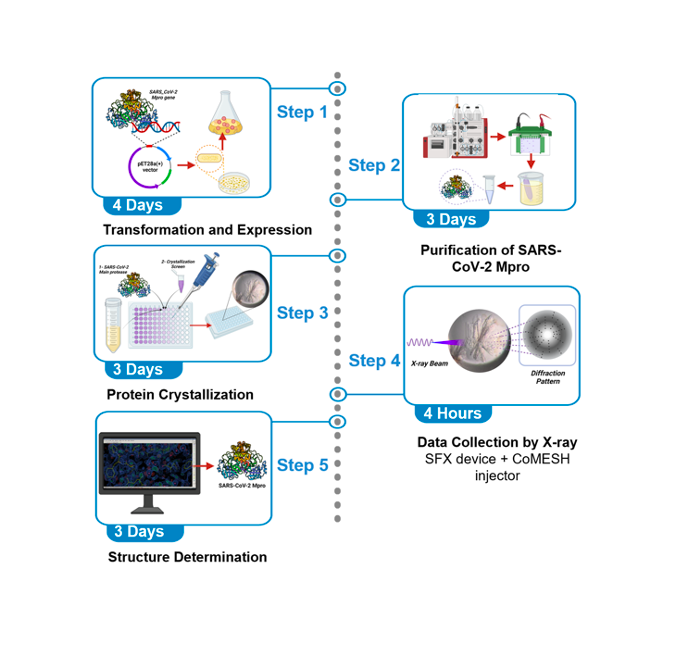
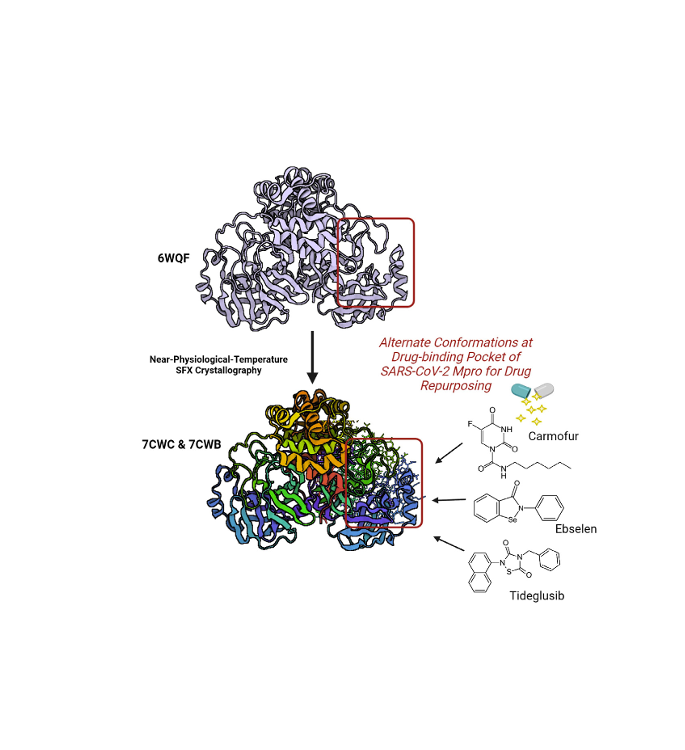
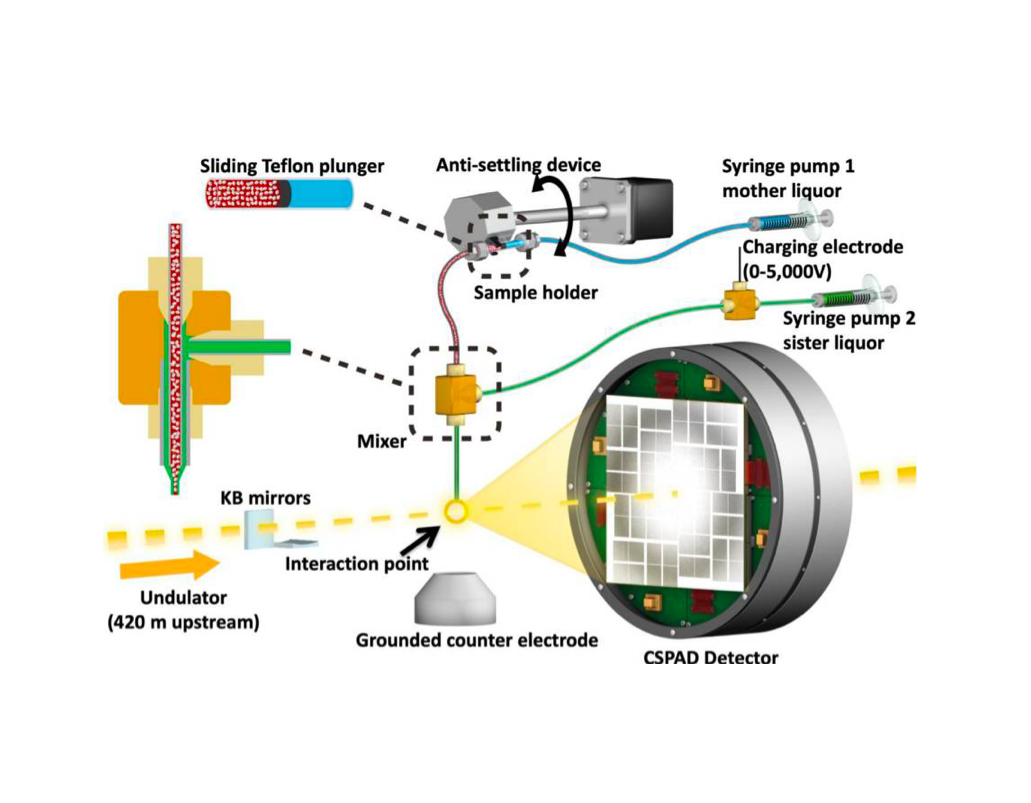
Severe acute respiratory syndrome coronavirus 2 main protease (SARS-CoV-2 Mpro) is a potential key drug target because of its essential role in this virus’ life cycle. Mpro mainly functions by processing the 1ab polyprotein. Inhibition of the Mpro activity with drugs is one of the main goals of the research community to tackle the COVID-19 pandemic. In this protocol, we report the steps to obtain high-throughput SARS-CoV-2 Mpro structures with a Serial Femtosecond X-ray crystallography (SFX) technique performed at XFEL. This protocol describes step-by-step high-resolution structure determination of the native SARS-CoV-2 Mpro at ambient temperature
![]()
, , , ,,, , , , , ,, , , , , , , , ,, , , , , ,
Nature Communications Biology 2022
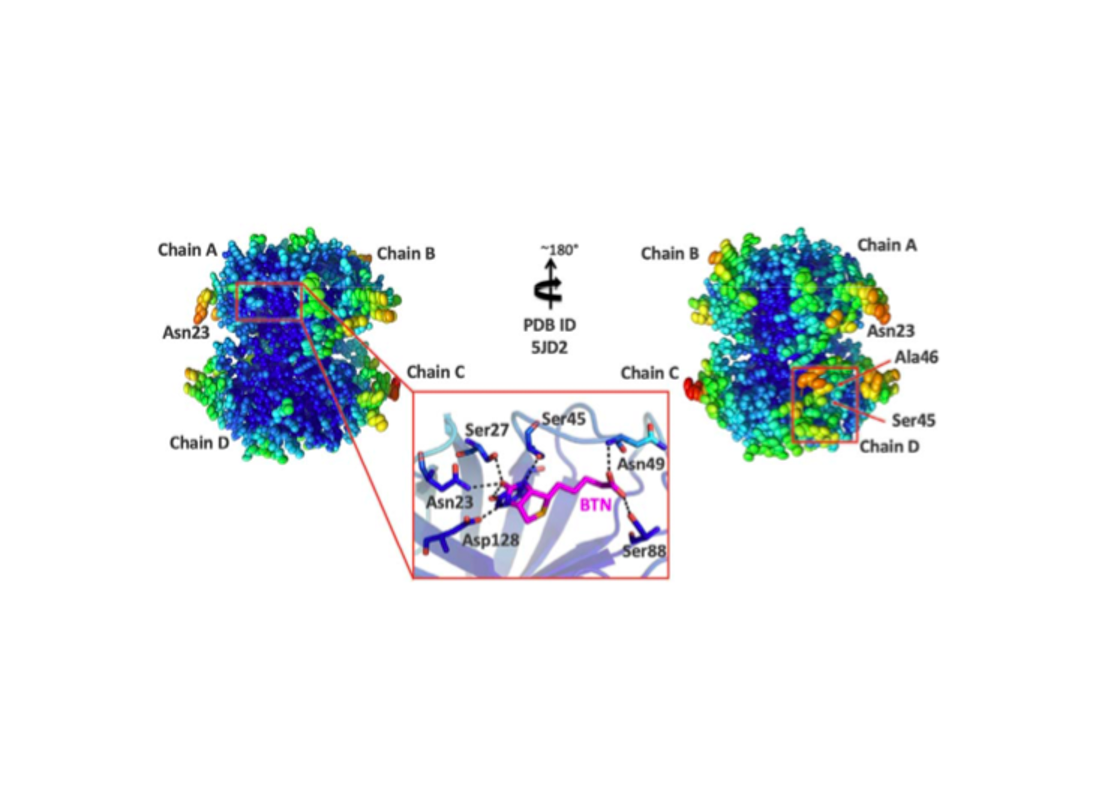
Multimeric protein assemblies are abundant in nature. Streptavidin is an attractive protein that provides a paradigm system to investigate the intra- and intermolecular interactions of multimeric protein complexes. Also, it offers a versatile tool for biotechnological applications. Here, we present two apo-streptavidin structures, the first one is an ambient temperature Serial Femtosecond X-ray crystal (Apo-SFX) structure at 1.7 Å resolution and the second one is a cryogenic crystal structure (Apo-Cryo) at 1.1 Å resolution. These structures are mostly in agreement with previous structural data. Combined with computational analysis, these structures provide invaluable information about structural dynamics of apo streptavidin. Collectively, these data further reveal a novel cooperative allostery of streptavidin which binds to substrate via water molecules that provide a polar interaction network and mimics the substrate biotin which displays one of the strongest affinities found in nature.
![]()
Crystals 2021
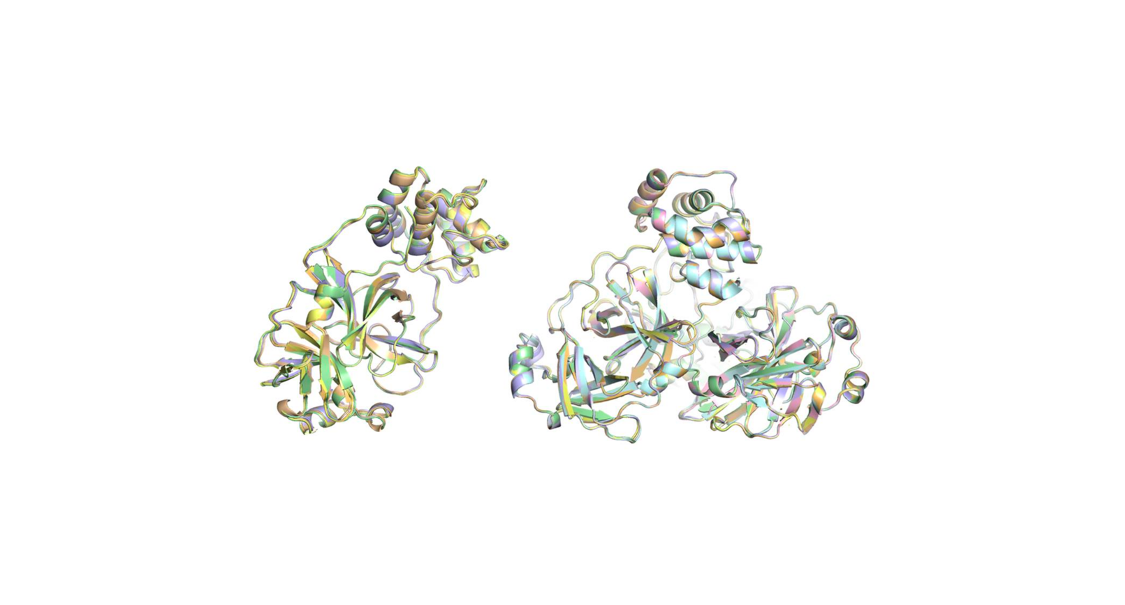
Since early 2020, COVID-19 has grown to affect the lives of billions globally. A worldwide investigation has been ongoing for characterizing the virus and also for finding an effective drug and developing vaccines. As time has been of the essence, a crucial part of this research has been drug repurposing; therefore, confirmation of in silico drug screening studies have been carried out for this purpose. Here we demonstrated the possibility of screening a variety of drugs efficiently by leveraging a high data collection rate of 120 images/second with the new low-noise, high dynamic range ePix10k2M Pixel Array Detector installed at the Macromolecular Femtosecond Crystallography (MFX) instrument at the Linac Coherent Light Source (LCLS). The X-ray Free-Electron Laser (XFEL) is used for remote high-throughput data collection for drug repurposing of the main protease (Mpro) of SARS-CoV-2 at ambient temperature with mitigated X-ray radiation damage. We obtained multiple structures soaked with nine drug candidate molecules in two crystal forms. Although our drug binding attempts failed, we successfully established a high-throughput Serial Femtosecond X-ray crystallographic (SFX) data collection protocol.
![]()
Ayan E. and DeMirci H.
2021
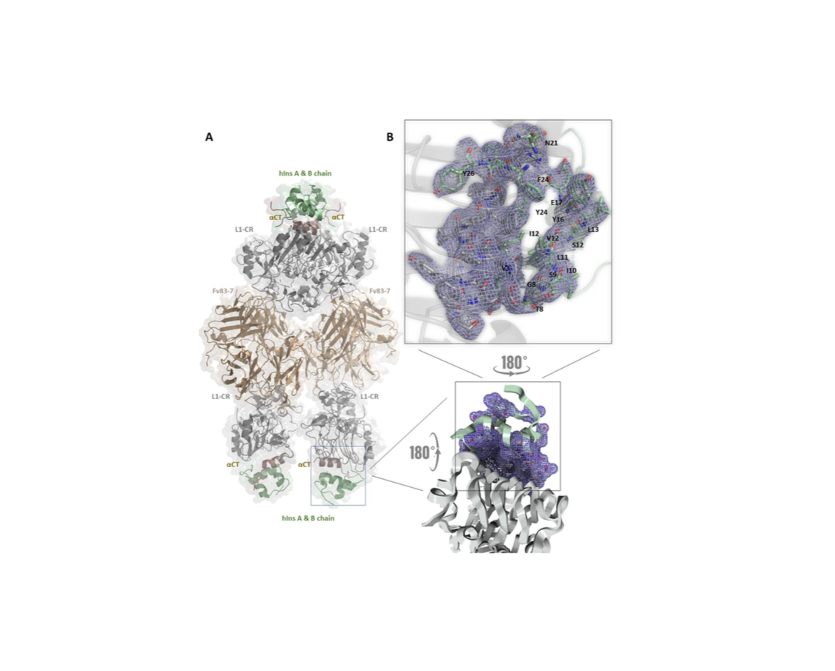
Insulin is an essential factor for the mammalian organisms: a regulator of glucose metabolism and other key signaling pathways. Insulin is also a multifunctional hormone whose absence can cause many diseases. Recombinant insulin is widely used in the treatment of diabetes. Understanding the insulin, biosimilars and biobetters from a holistic perspective will help pharmacologically user-friendly molecule design and develop personalized medicine- oriented therapeutic strategies for diabetes. Additionally, it helps to understand the underlying mechanism of other insulin-dependent metabolic disorders. The purpose of this atlas is to review insulin from a biotechnological, basic science, clinical perspective; explain nearly all insulin- related disorders and their underlying molecular mechanisms; explore exogenous/recombinant production strategies of patented and research-level insulin/analogs; and highlight their mechanism of action from a structural perspectives. Combined with computational analysis, comparisons of the insulin and analogs also provide novel information about the structural dynamics of the insulin.
![]()
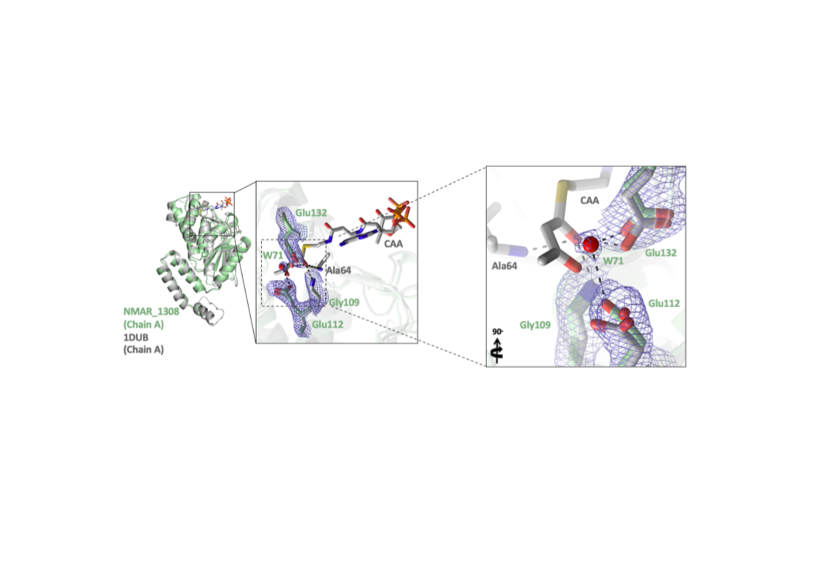
Destan E., Yuksel B., Tolar B.B., Ayan E., Deutsch S., Yoshikuni Y., Wakatsuki S., Francis C.A., DeMirci H.
Scientific Reports 2021
The ammonia-oxidizing thaumarchaeal 3-hydroxypropionate/4-hydroxybutyrate (3HP/4HB) cycle is one of the most energy-efficient CO2 fixation cycles discovered thus far. The protein encoded by Nmar_1308 (from Nitrosopumilus maritimus SCM1) is a promiscuous enzyme that catalyzes two essential reactions within the thaumarchaeal 3HP/4HB cycle, functioning as both a crotonyl-CoA hydratase (CCAH) and 3- hydroxypropionyl-CoA dehydratase (3HPD). In performing both hydratase and dehydratase activities, Nmar_1308 reduces the total number of enzymes necessary for CO2 fixation in Thaumarchaeota, reducing the overall cost for biosynthesis. Here, we present the first high-resolution crystal structure of this bifunctional enzyme with key catalytic residues in the thaumarchaeal 3HP/4HB pathway.
![]()

Tanisali G., Sokak A., Bulut A.S., Sander T.Z., Dogan O., Dag C., Gonen M., Can F., DeMirci H., Ergonul O.
International Journal of Infectious Diseases 2021
We aimed to compare the effectiveness of different masks in limiting the dispersion of coughed air. We employed the single-curved mirror Schlieren method. Coughed air with a slightly higher temperature than the ambient air generates a refractive index gradient. We used a spherical mirror with a radius of curvature of 10 m and a diameter of 60 cm. The spread of the cough wavefront was investigated among five subjects wearing (1) no mask, (2) single surgical mask, (3) double surgical masks, (4) cloth mask, 5) valveless N95 mask or 6) valved N95 mask. All mask types significantly reduced the magnitude of the dispersion of the contaminated region. The percent reduction in the cross sectional area of the contaminated region for the same mask types but different subjects revealed by the normalized data suggests that the fitting of the masks play an important role. We observed no significant difference between using either a single or a double surgical mask for the exhalation spread. Cloth masks could be effective, depending on the quality of the cloth. The valved-N95 mask type exclusively protects the user. Fitting of the mask is an important factor to minimize the contaminated region.
![]()
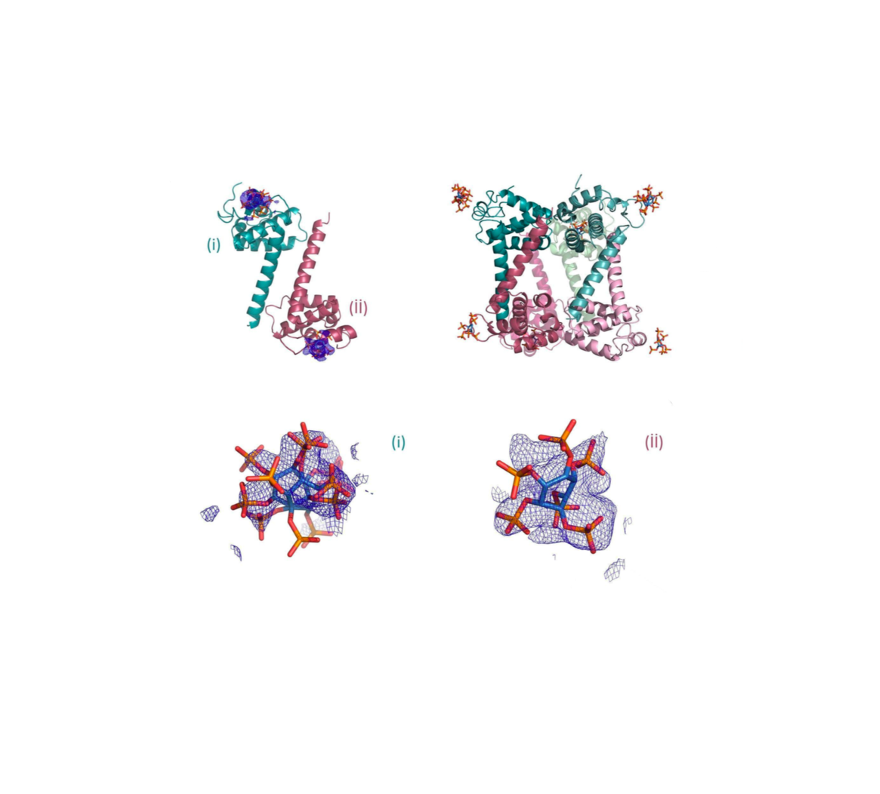
, , , ,, , , , ,, , , , , , , , , , , , , , , , ,
Scientific Reports 2021
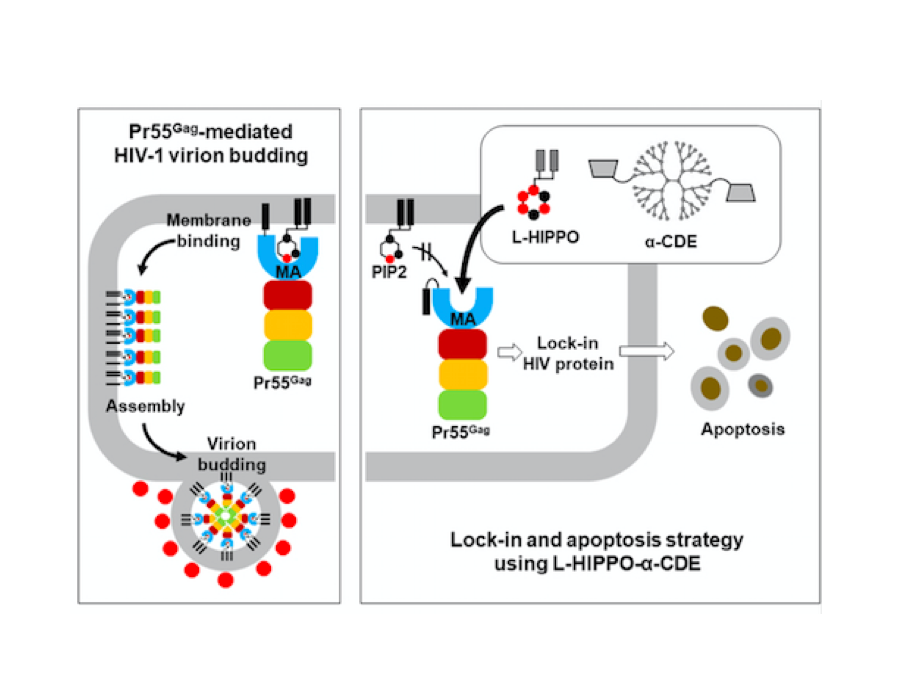
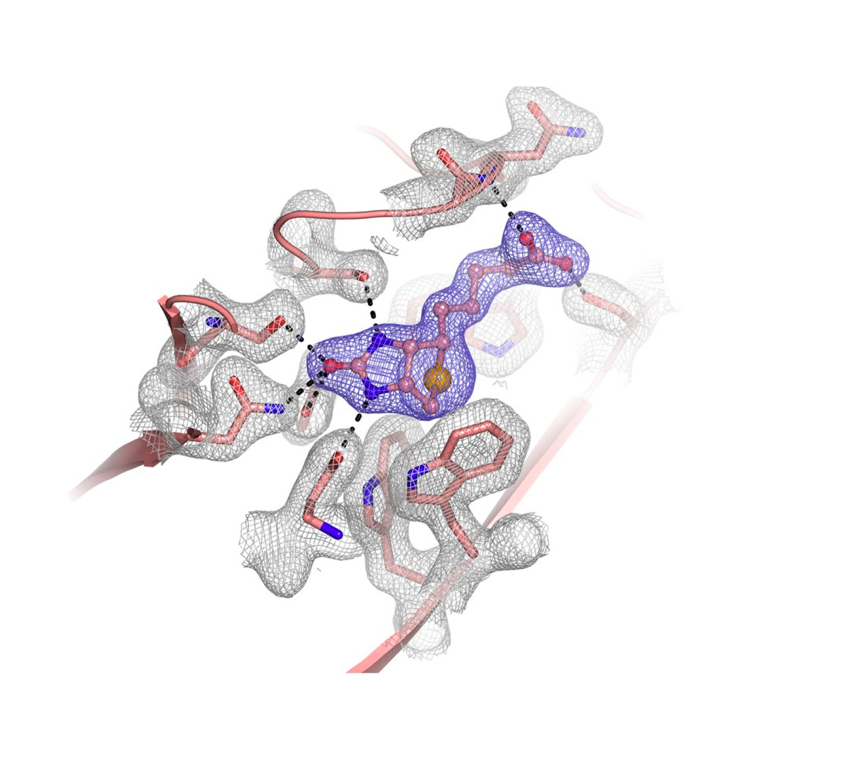
Oligomerization of Pr55Gag is a critical step of the late stage of the HIV life cycle. It has been known that the binding of IP6, an abundant endogenous cyclitol molecule at the MA domain, has been linked to the oligomerization of Pr55Gag. However, the exact binding site of IP6 on MA remains unknown and the structural details of this interaction are missing. Here, we present three high‐resolution crystal structures of the MA domain in complex with IP6 molecules to reveal its binding mode. Additionally, extensive Differential Scanning Fluorimetry analysis combined with cryo‐ and ambient‐ temperature X‐ray crystallography and GNM‐based transfer entropy calculations identify the key residues that participate in IP6 binding. Our data provide novel insights about the multilayered HIV‐1 virion assembly process that involves the interplay of IP6 with PIP2, a phosphoinositide essential for the binding of Pr55Gag to membrane. IP6 and PIP2 have neighboring alternate binding sites within the same highly basic region (residues 18–33). This indicates that IP6 and PIP2 bindings are not mutually exclusive and may play a key role in coordinating virion particles’ membrane localization. Based on our three different IP6‐MA complex crystal structures, we propose a new model that involves IP6 coordination of the oligomerization of outer MA and inner CA domain’s 2D layers during assembly and budding.
![]()
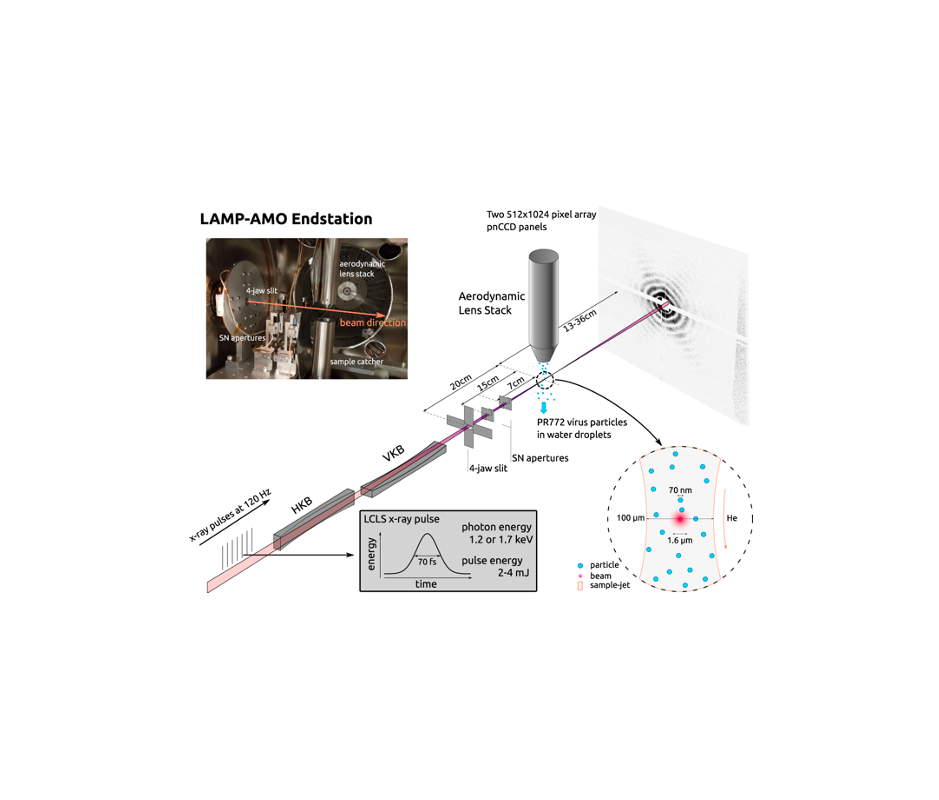
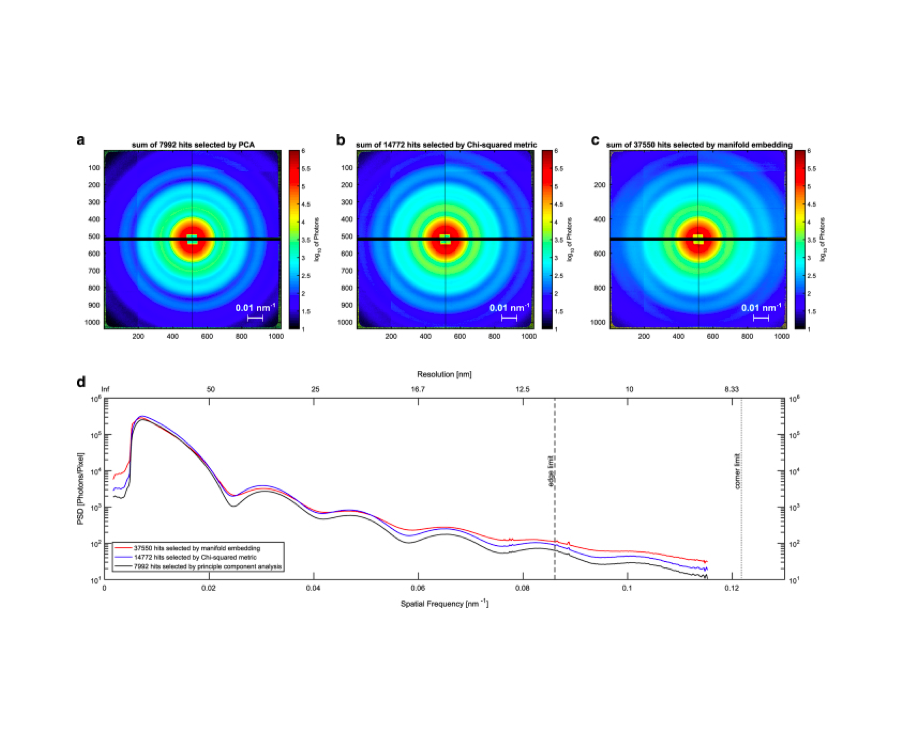
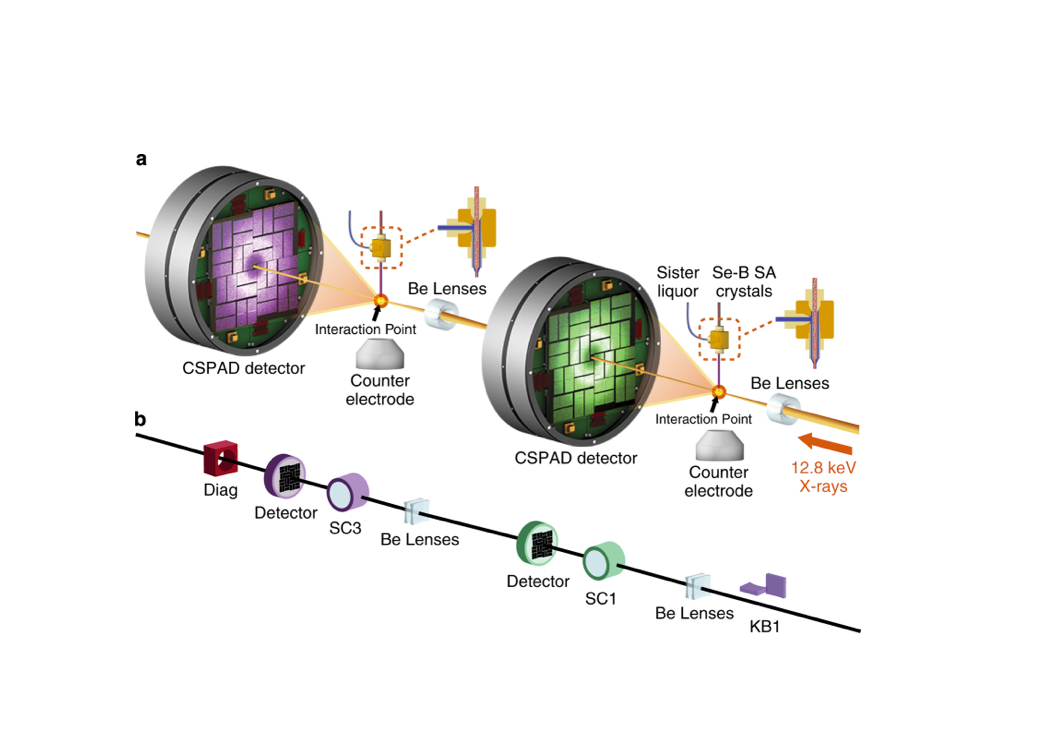
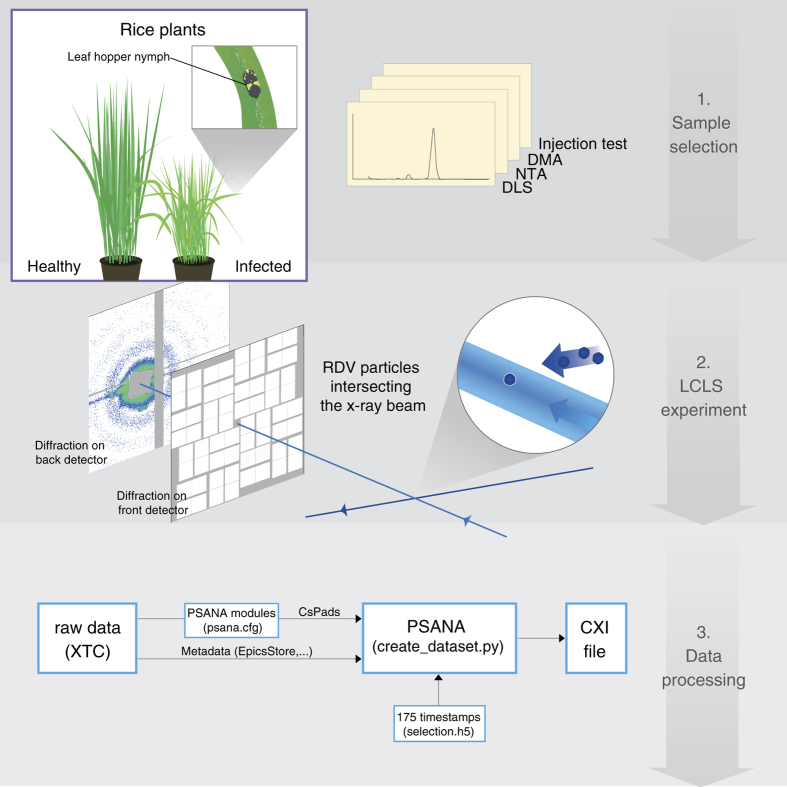
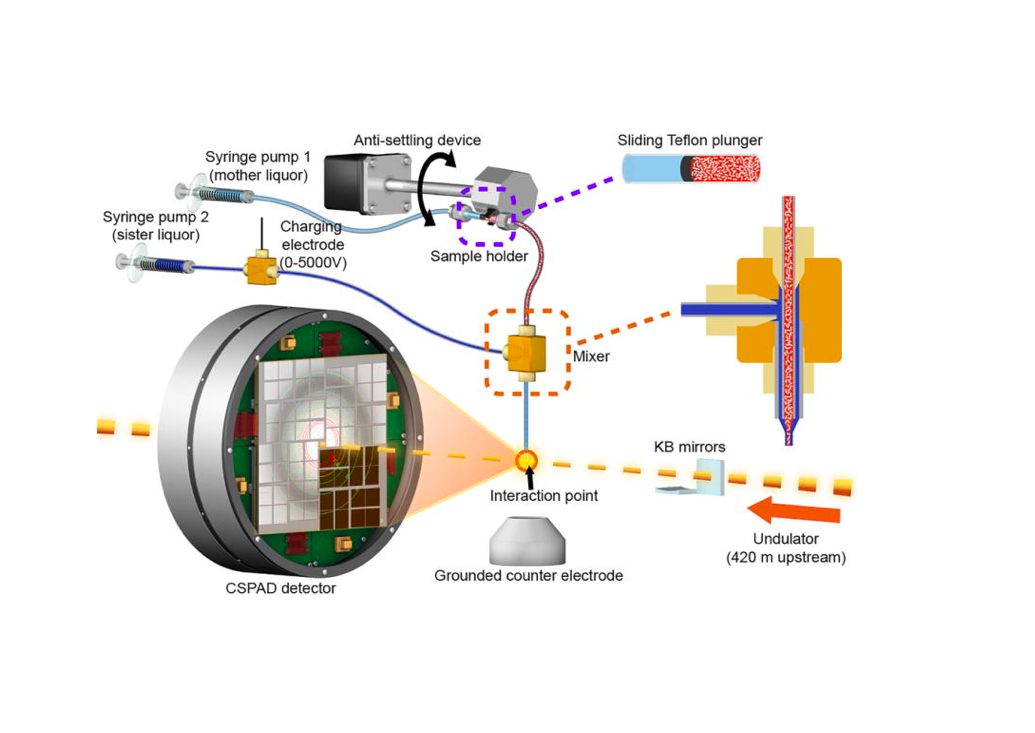
Single Particle Imaging (SPI) with intense coherent X-ray pulses from X-ray free-electron lasers (XFELs) has the potential to produce molecular structures without the need for crystallization or freezing. Here we present a dataset of 285,944 diffraction patterns from aerosolized Coliphage PR772 virus particles injected into the femtosecond X-ray pulses of the Linac Coherent Light Source (LCLS). Additional exposures with background information are also deposited. The diffraction data were collected at the Atomic, Molecular and Optical Science Instrument (AMO) of the LCLS in 4 experimental beam times during a period of four years. The photon energy was either 1.2 or 1.7 keV and the pulse energy was between 2 and 4 mJ in a focal spot of about 1.3 μm x 1.7 μm full width at half maximum (FWHM). The X-ray laser pulses captured the particles in random orientations. The data offer insight into aerosolised virus particles in the gas phase, contain information relevant to improving experimental parameters, and provide a basis for developing algorithms for image analysis and reconstruction.![]()
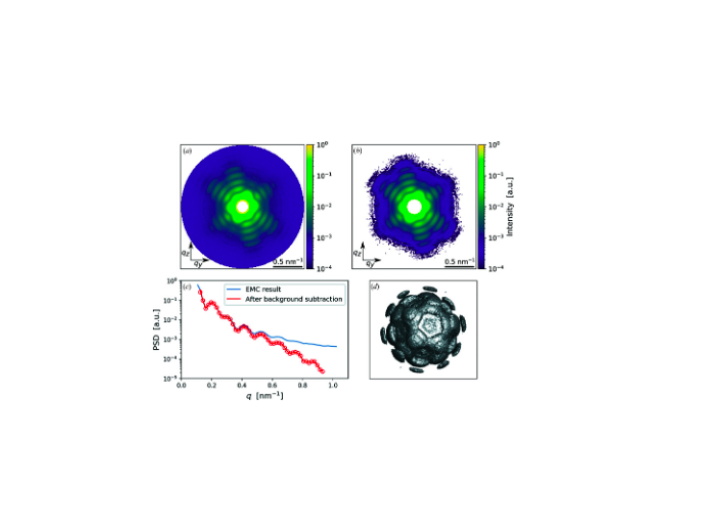
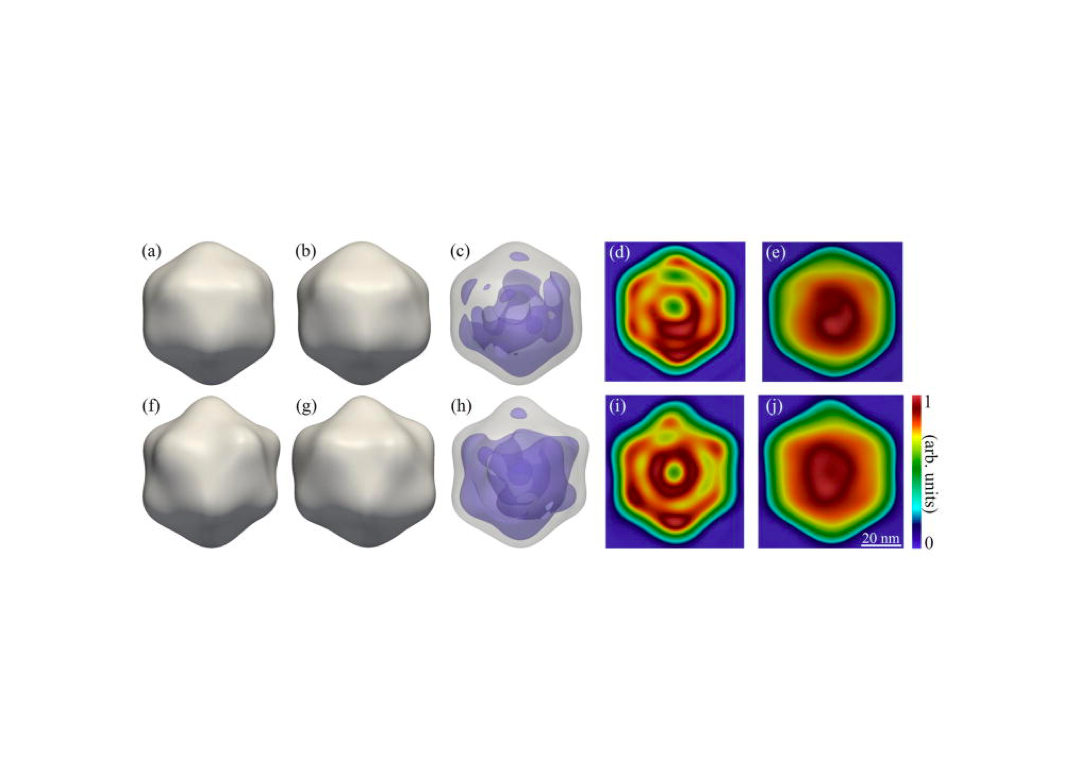
An improved analysis for single-particle imaging (SPI) experiments, using the limited data, is presented here. Results are based on a study of bacteriophage PR772 performed at the Atomic, Molecular and Optical Science instrument at the Linac Coherent Light Source as part of the SPI initiative. Existing methods were modified to cope with the shortcomings of the experimental data: inaccessibility of information from half of the detector and a small fraction of single hits. The general SPI analysis workflow was upgraded with the expectation-maximization based classification of diffraction patterns and mode decomposition on the final virus-structure determination step. The presented processing pipeline allowed us to determine the 3D structure of bacteriophage PR772 without symmetry constraints with a spatial resolution of 6.9 nm. The obtained resolution was limited by the scattering intensity during the experiment and the relatively small number of single hits. PubMed![]()
Durdagi, S., Avsar, T., Orhan, M. D., Serhatli, M., Balcioglu, B. K., Ozturk, H. U., Kayabolen, A., Cetin, Y., Aydinlik, S., Bagci-Onder, T., Tekin, S., Demirci, H., Guzel, M., Akdemir, A., Calis, S., Oktay, L., Tolu, I., Butun, Y. E., Erdemoglu, E., Olkan, A., Tokay, N., Işik, Ş., Ozcan, A., Acar, E., Buyukkilic, S. And Yumak, Y.
2020
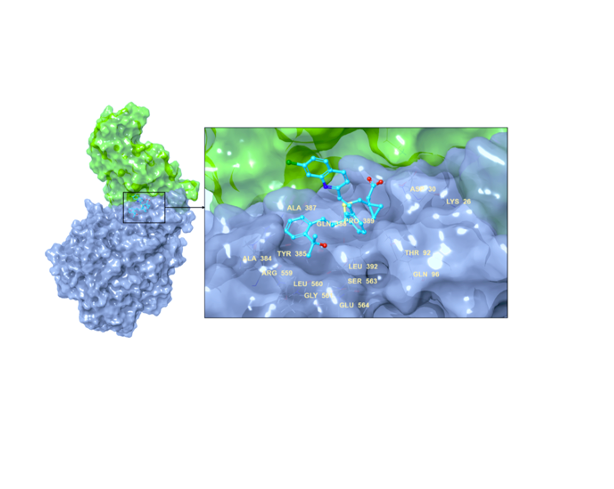
Small molecule inhibitors have previously been investigated in different studies as possible therapeutics in the treatment of SARS-CoV-2. In the current drug repurposing study, we identified the leukotriene (D4) receptor antagonist Montelukast as a novel agent that simultaneously targets two important drug targets of SARS-CoV-2. We initially demonstrated the dual inhibition profile of Montelukast through multiscale molecular modeling studies. Next, we characterized its effect on both targets by different in vitro experiments including the Fluorescent Resonance Energy Transfer (FRET)-based main protease enzyme inhibition assay, surface plasmon resonance (SPR) spectroscopy, pseudovirus neutralization on HEK293T / hACE2, and virus neutralization assay using xCELLigence MP real time cell analyzer. Our integrated in silico and in vitro results confirmed the dual potential effect of the Montelukast both on virus entry into the host cell (Spike/ACE2) and on the main protease enzyme inhibition. The virus neutralization assay results showed that while no cytotoxicity of the Montelukast was observed at 12 μM concentration, the cell index time 50 (CIT50) value was delayed for 12 hours. Moreover, it was also shown that Favipiravir, a well-known antiviral used in COVID-19 therapy, should be used by 16-fold higher concentrations than Montelukast in order to have the same effect of Montelukast. The rapid use of new small molecules in the pandemic is very important today. Montelukast, whose pharmacokinetic and pharmacodynamic properties are very well characterized and has been widely used in the treatment of asthma since 1998, should urgently be completed in clinical phase studies and if its effect is proven in clinical phase studies, it should be used against COVID-19.
![]()
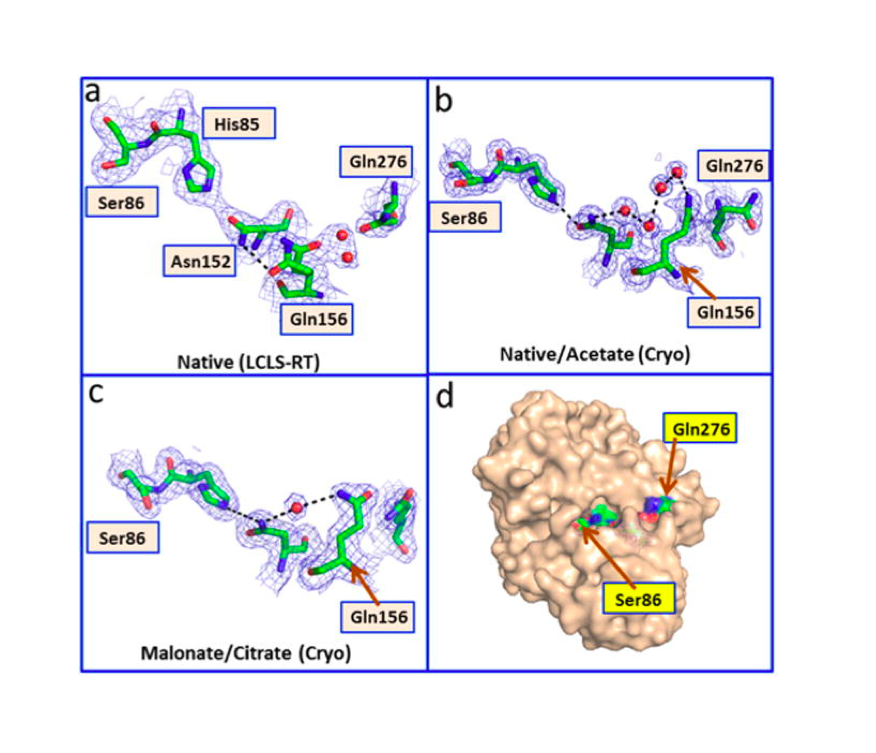
The COVID19 pandemic has resulted in 25+ million reported infections and nearly 850.000 deaths. Research to identify effective therapies for COVID19 includes: i) designing a vaccine as future protection; ii) structure-based drug design; and iii) identifying existing drugs to repurpose them as effective and immediate treatments. To assist in drug repurposing and design, we determined two apo structures of Severe Acute Respiratory Syndrome CoronaVirus-2 main protease at ambient-temperature by Serial Femtosecond X-ray crystallography. We employed detailed molecular simulations of selected known main protease inhibitors with the structures and compared binding modes and energies. The combined structural biology and molecular modeling studies not only reveal the dynamics of small molecules targeting main protease but will also provide invaluable opportunities for drug repurposing and structure-based drug design studies against SARS-CoV-2.
![]()
![]()
Autotrophic microorganisms that convert inorganic carbon into organic matter were key players in the evolution of life on early Earth. As the early atmosphere became oxygenated, these microorganisms needed protection from oxygen, which was especially important for those organisms that relied on enzymes with oxygen-sensitive metal clusters (e.g., Fe-S). Here we investigated how the key enzyme of the 3-hydroxypropionate/4-hydroxybutyrate (HP/HB) cycle for CO2-fixation, 4-hydroxybutyryl-CoA dehydratase (4HBD), adapted from anoxic to oxic conditions. 4HBD is found in both anaerobic bacteria and aerobic ammonia-oxidizing archaea (AOA). The oxygen-sensitive bacterial 4HBD and oxygen-tolerant archaeal 4HBD share 59 % amino acid identity. To examine the structural basis of oxygen tolerance in archaeal 4HBD, we determined the atomic resolution structure of the enzyme. Two tunnels providing access to the canonical 4Fe-4S cluster in oxygen-sensitive bacterial 4HBD were closed with four conserved mutations found in all aerobic AOA and other archaea. Further biochemical experiments support our findings that restricting access to the active site is key to oxygen tolerance, explaining how active site evolution drove a major evolutionary transition.
![]()
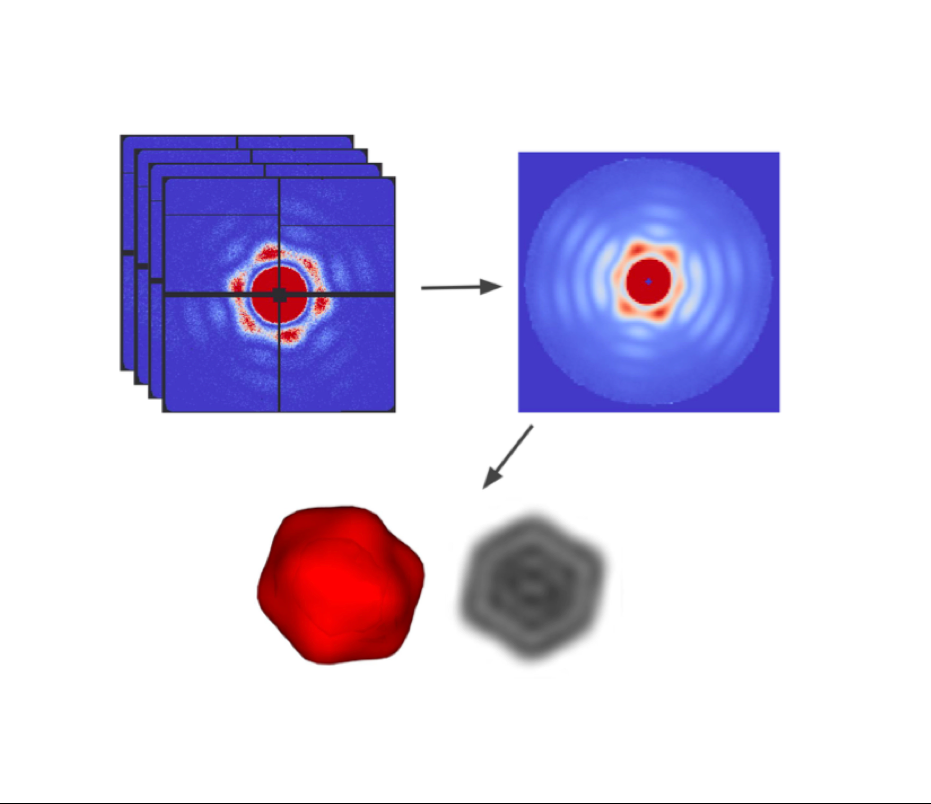
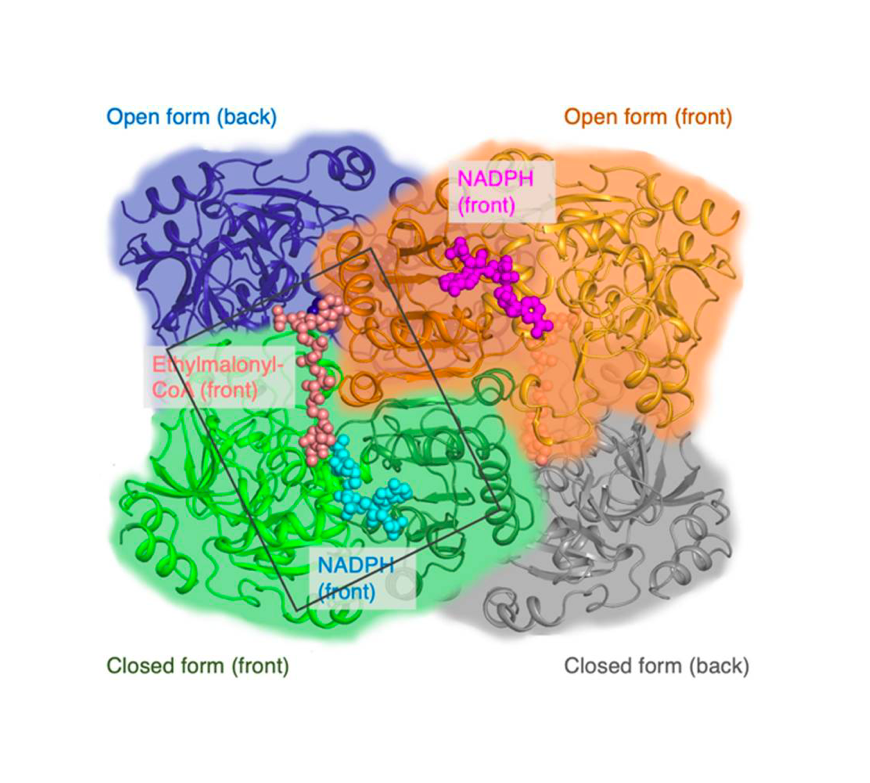
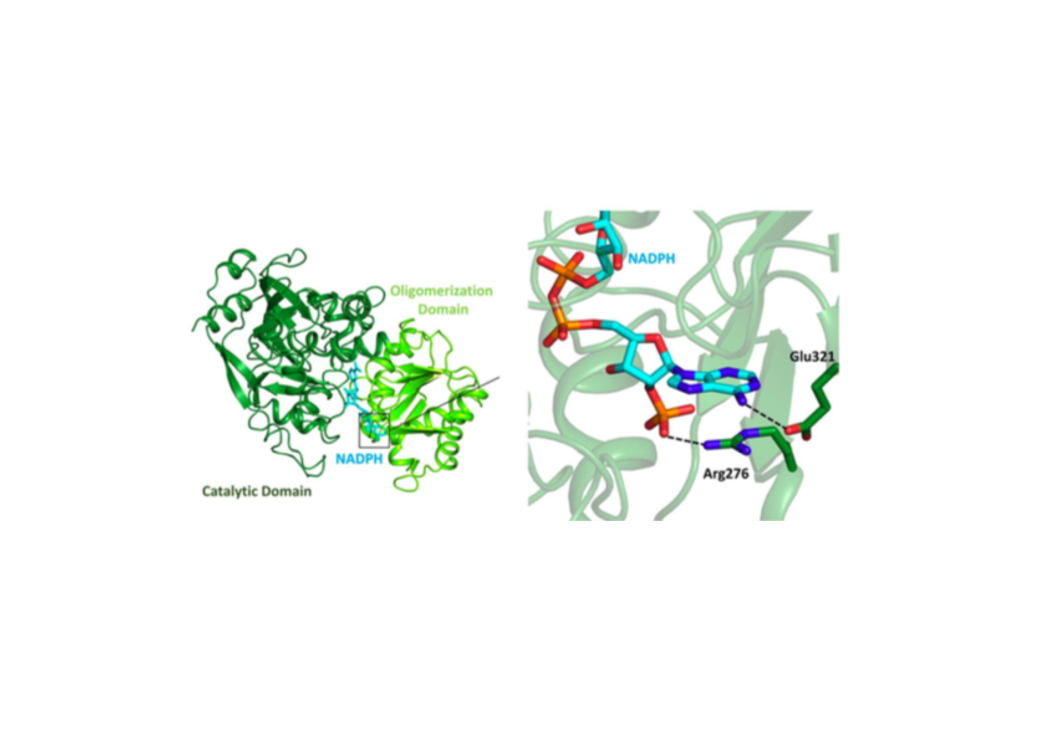
Enoyl-CoA carboxylases/reductases (ECRs) are the most efficient CO2-fixing enzymes described to date, outcompeting RubisCO, the key enzyme in photosynthesis in catalytic activity by more than an order of magnitude. However, the molecular mechanisms underlying ECR’s extraordinary catalytic activity remain elusive. Here we used different crystallographic approaches, including ambient temperature X-ray Free Electron Laser (XFEL) experiments, to study the dynamic structural organization of the ECR from Kitasatospora setae. K. setaeECR is a homotetramer that differentiates into a dimer of dimers of open- and closed-form subunits in the catalytically active state, suggesting that the enzyme operates with “half-site reactivity” to achieve high catalytic rates. Using structure-based mutagenesis, we show that catalysis is synchronized in K. setae ECR across the pair of dimers by conformational coupling of catalytic domains and within individual dimers by shared substrate binding sites. Our results provide unprecedented insights into the dynamic organization and synchronized inter- and intra-subunit communications of nature’s most efficient CO2-fixing enzyme during catalysis.
![]()
Int J Mol Sci. 2019
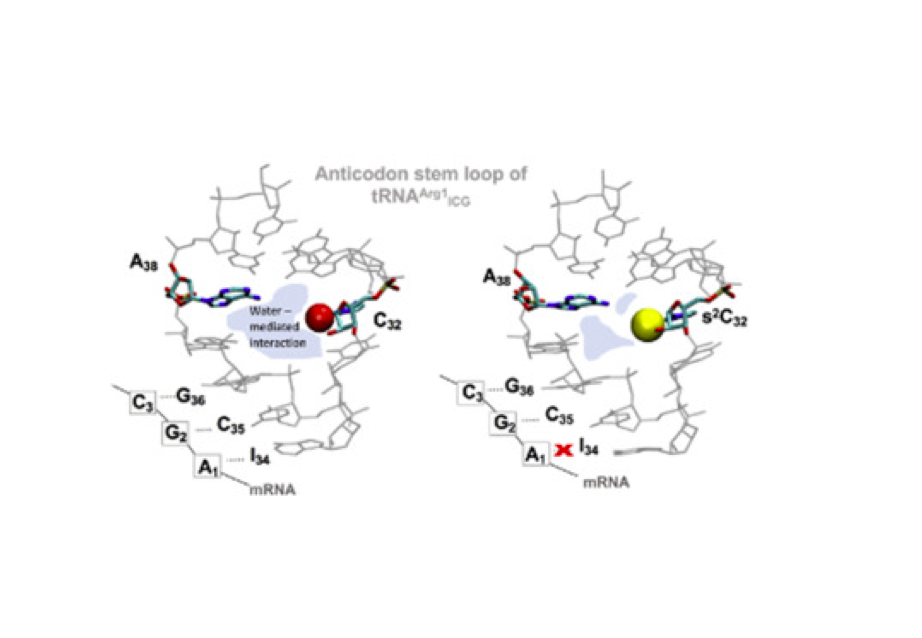
The Human immunodeficiency virus-1 (HIV-1) matrix (MA) domain is involved in the highly regulated assembly process of the virus particles that occur at the host cell’s plasma membrane. High-resolution structures of the MA domain determined using cryo X-ray crystallography have provided initial insights into the possible steps in the viral assembly process. However, these structural studies have relied on large and frozen crystals in order to reduce radiation damage caused by the intense X-rays. Here, we report the first X-ray free-electron laser (XFEL) study of the HIV-1 MA domain’s interaction with inositol hexaphosphate (IP6), a phospholipid headgroup mimic. We also describe the purification, characterization and micro crystallization of two MA crystal forms obtained in the presence of IP6. In addition, we describe the capabilities of serial femtosecond X-ray crystallography (SFX) using an XFEL to elucidate the diffraction data of MA-IP6 complex microcrystals in liquid suspension at ambient temperature. Two different microcrystal forms of the MA-IP6 complex both diffracted to beyond 3.5 Å resolution, demonstrating the feasibility of using SFX to study the complexes of MA domain of HIV-1 Gag polyprotein with IP6 at near-physiological temperatures. Further optimization of the experimental and data analysis procedures will lead to better understanding of the MA domain of HIV-1 Gag and IP6 interaction at high resolution and will provide basis for optimization of the lead compounds for efficient inhibition of the Gag protein recruitment to the plasma membrane prior to virion formation. PubMed
![]()
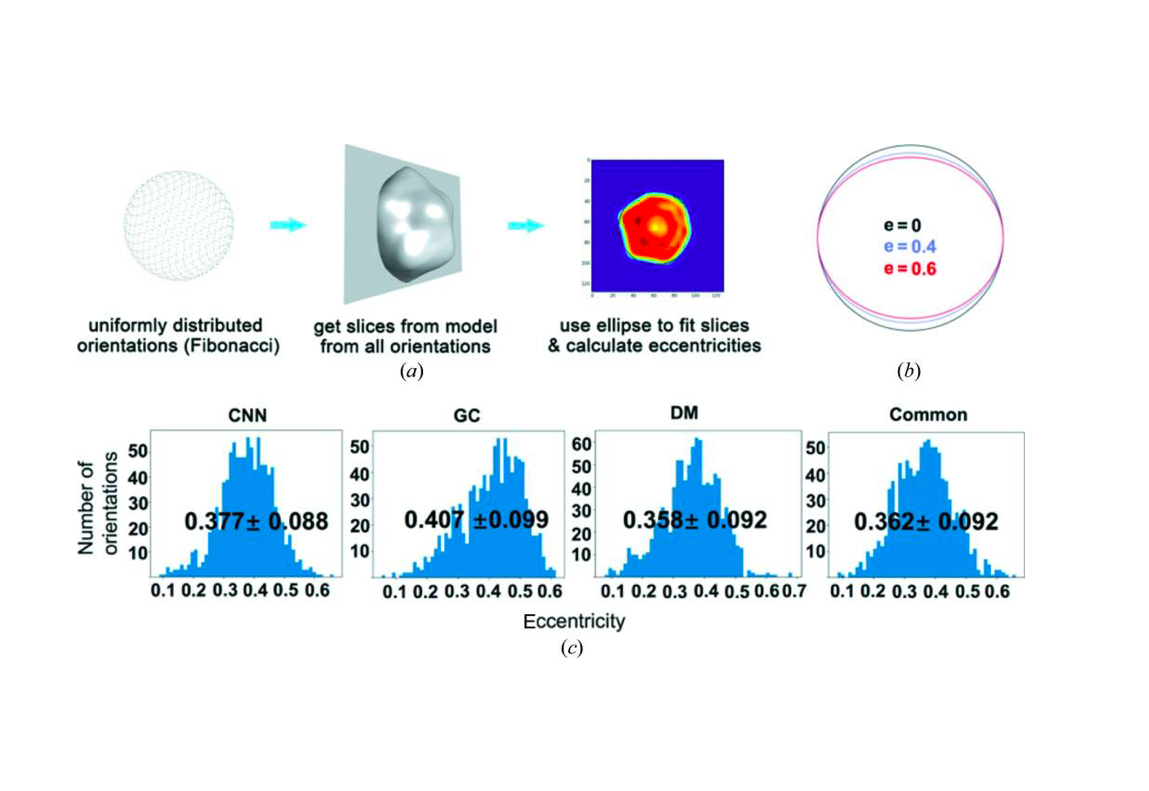
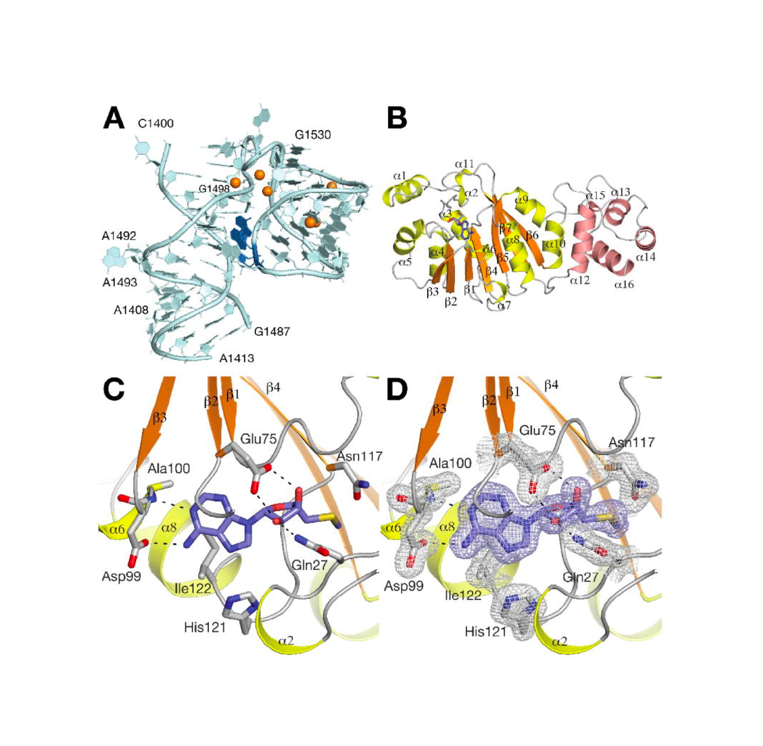
IUCrJ. 2019
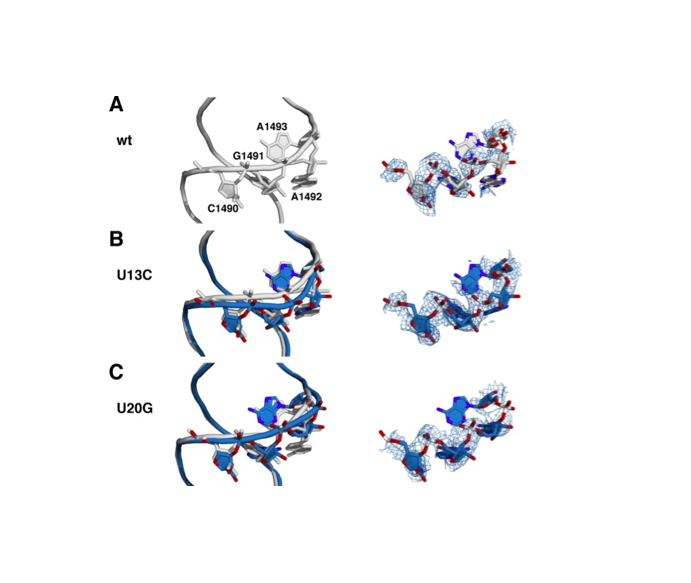
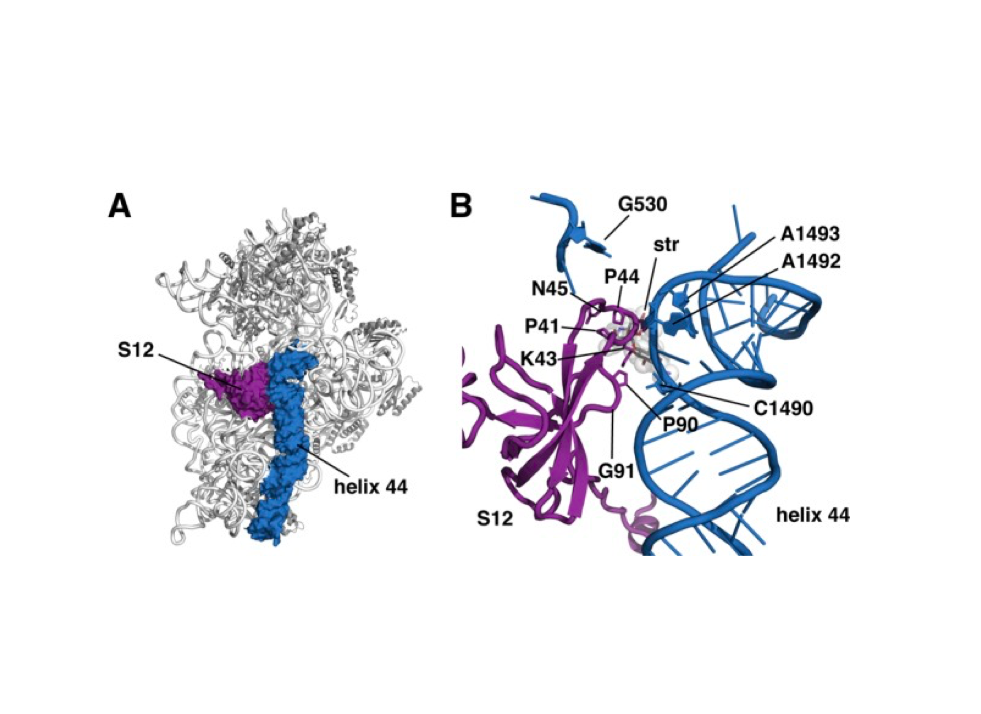
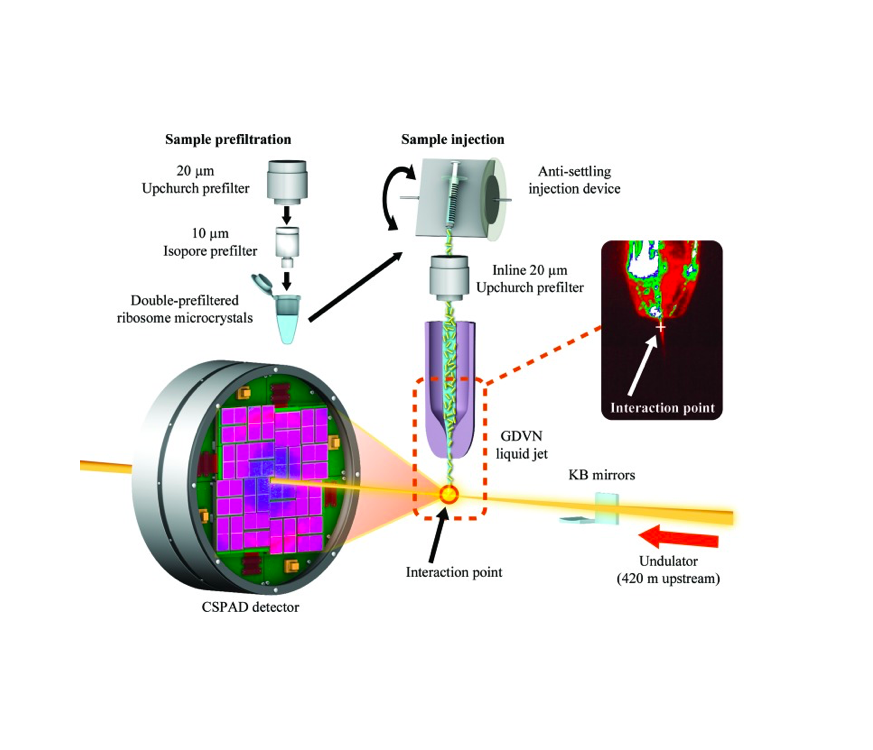
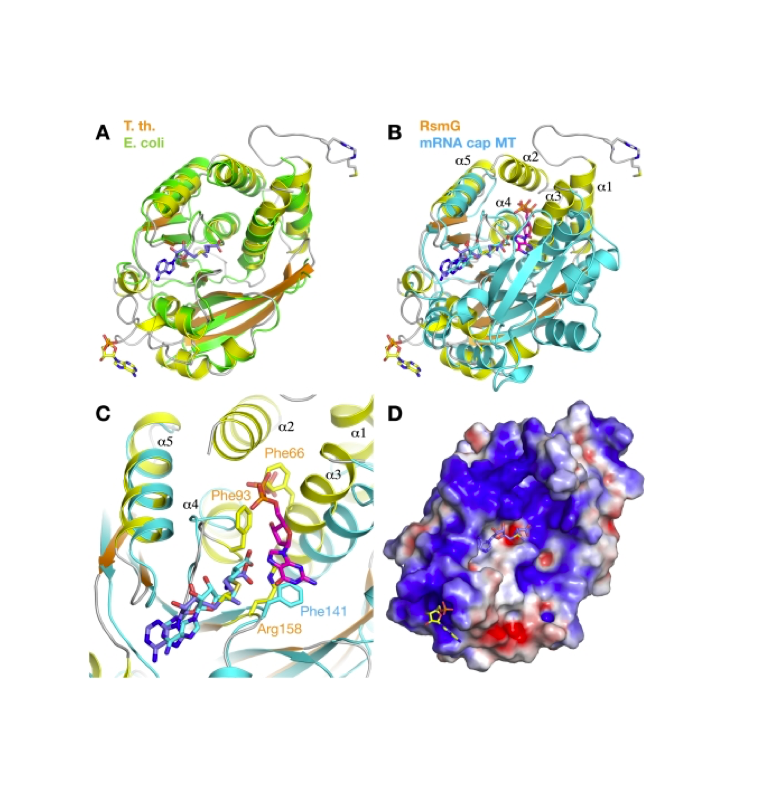
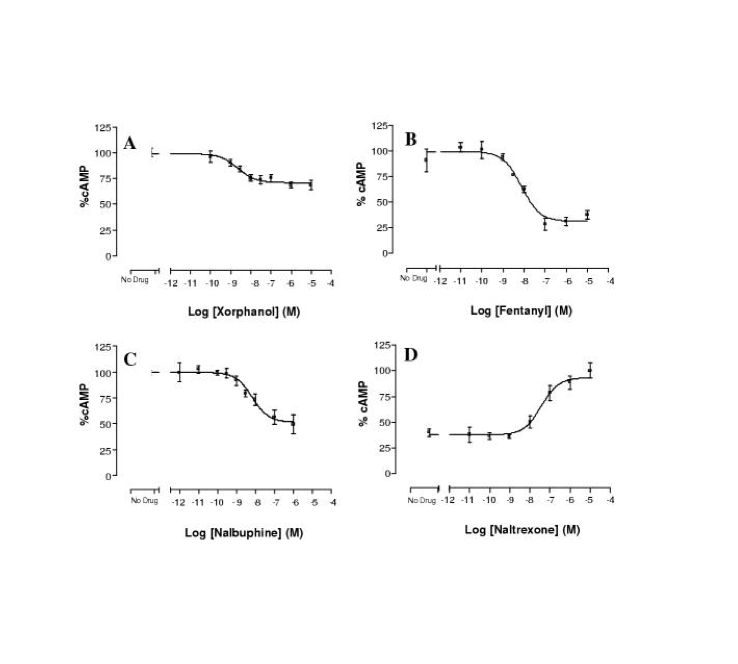
J Synchrotron Radiat. 2019
![]()
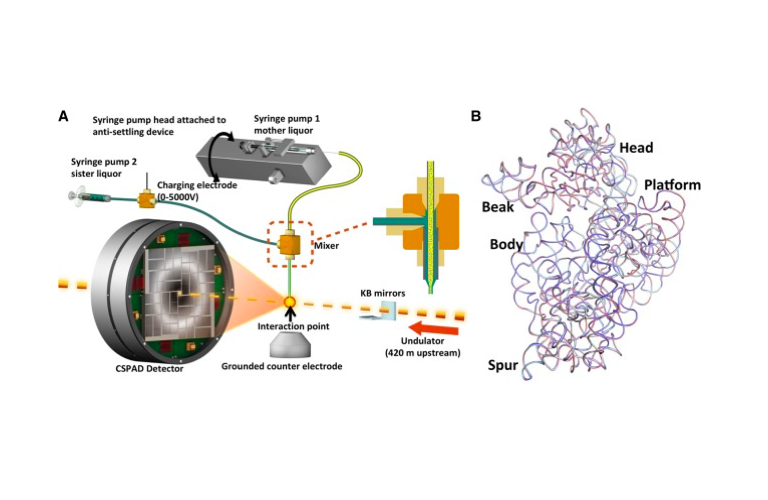
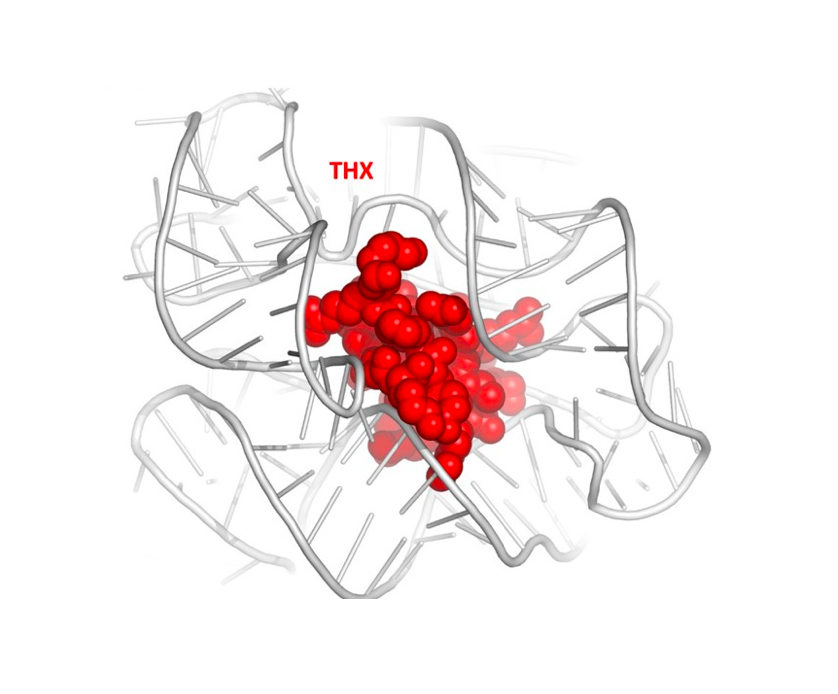
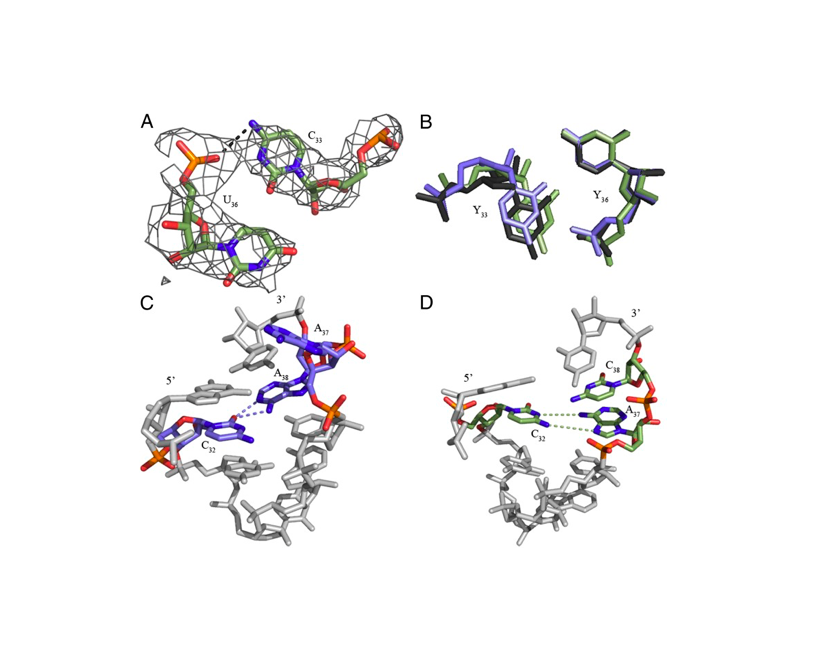
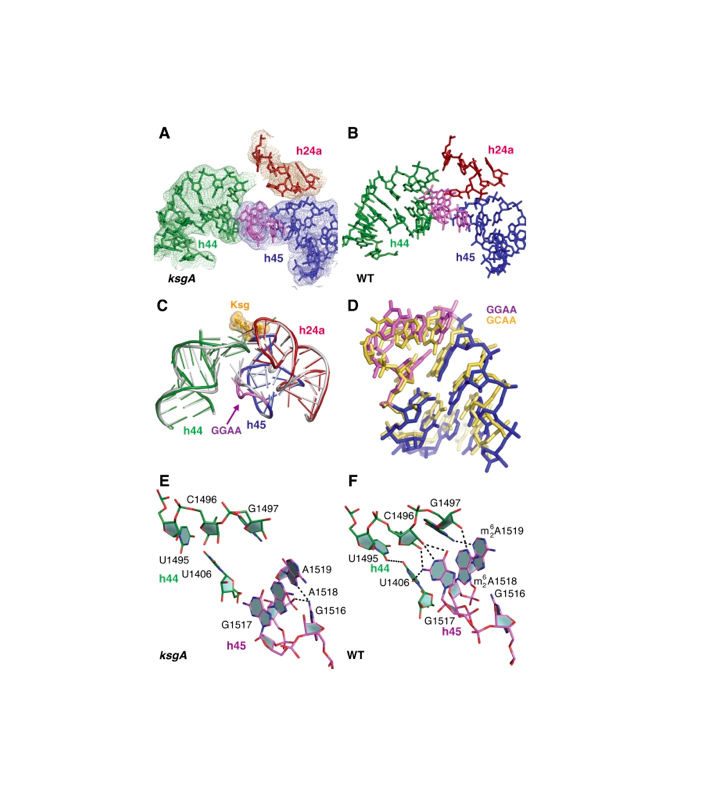
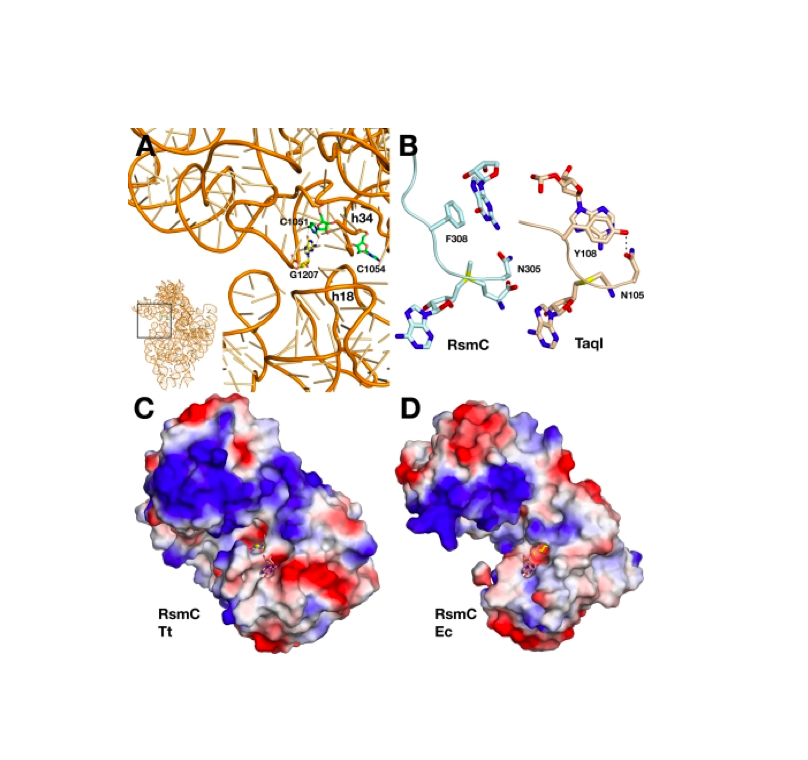
RNA 2018
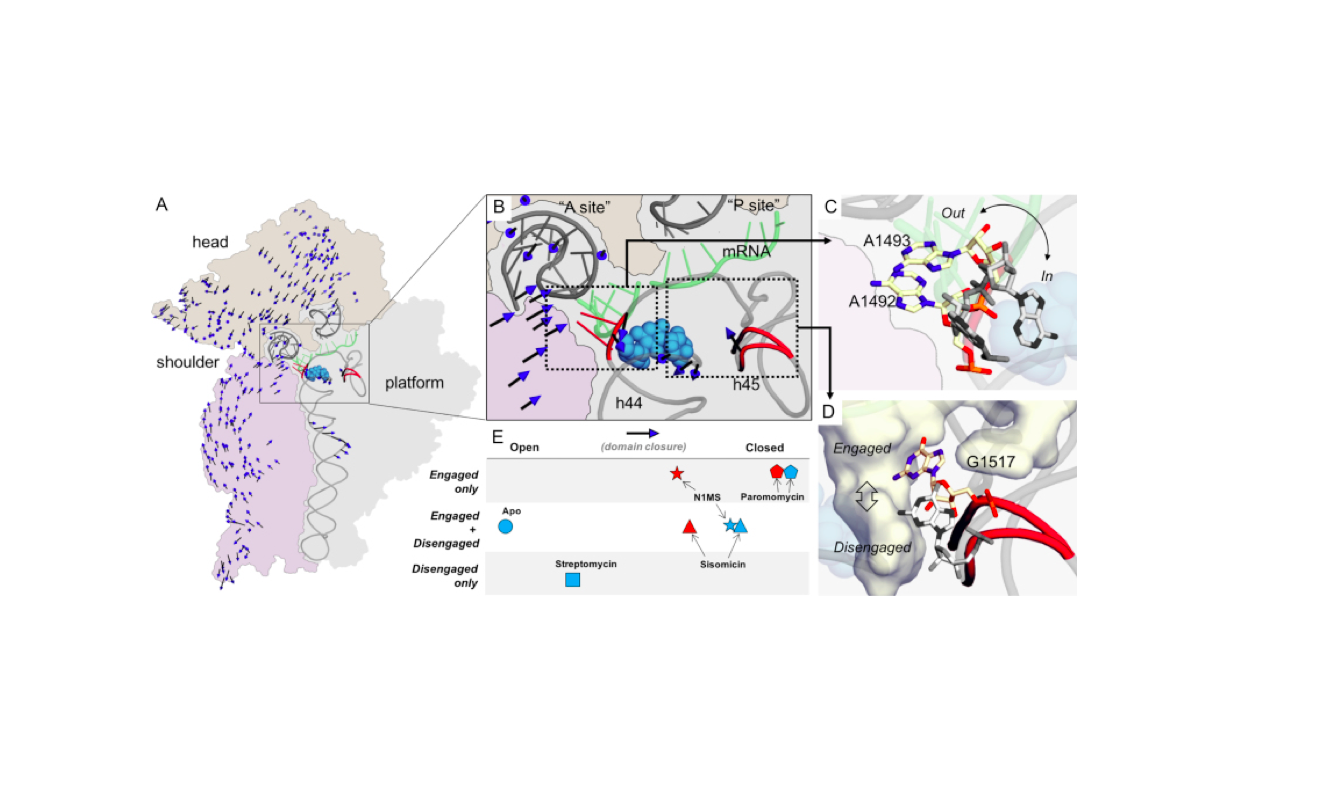
Nucleic Acids Res. 2018
![]()
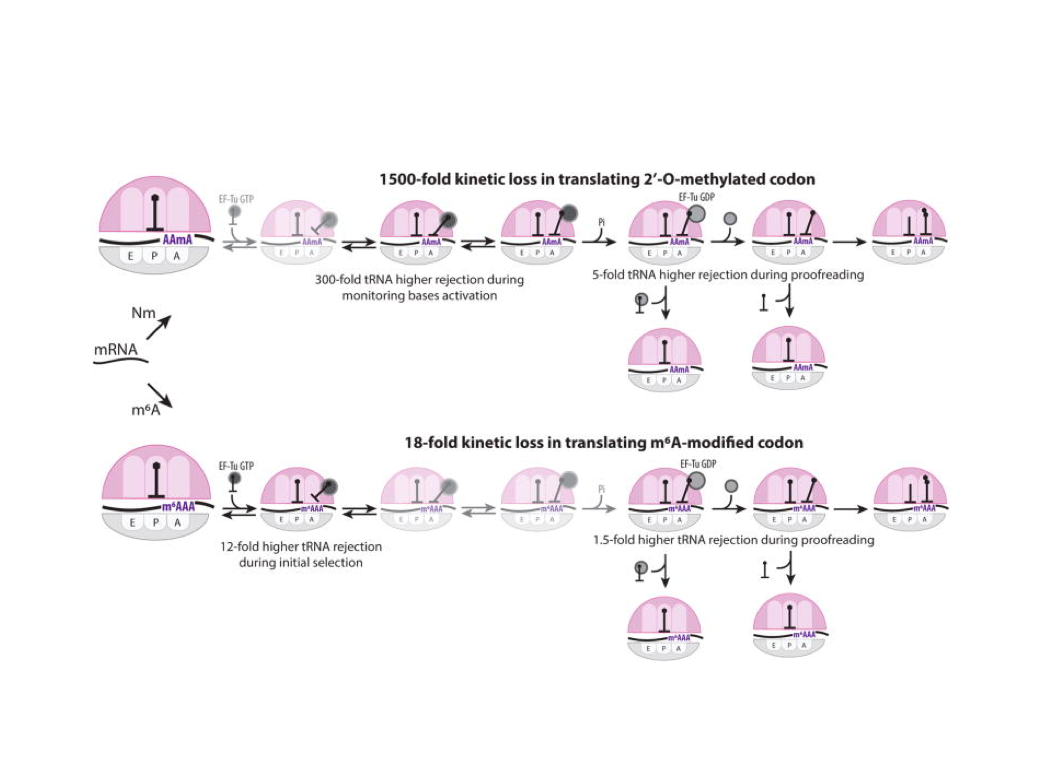
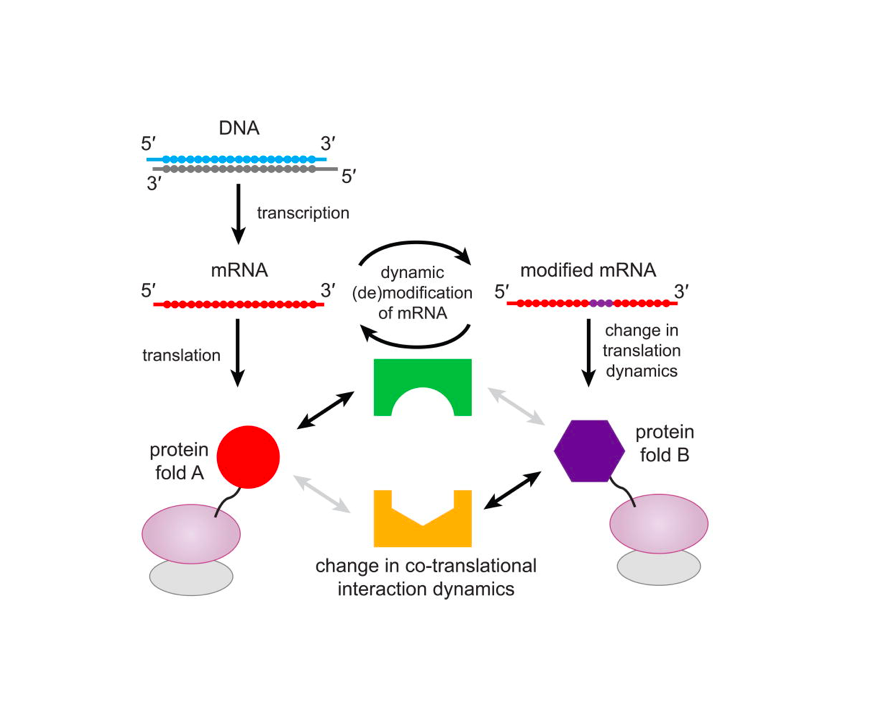
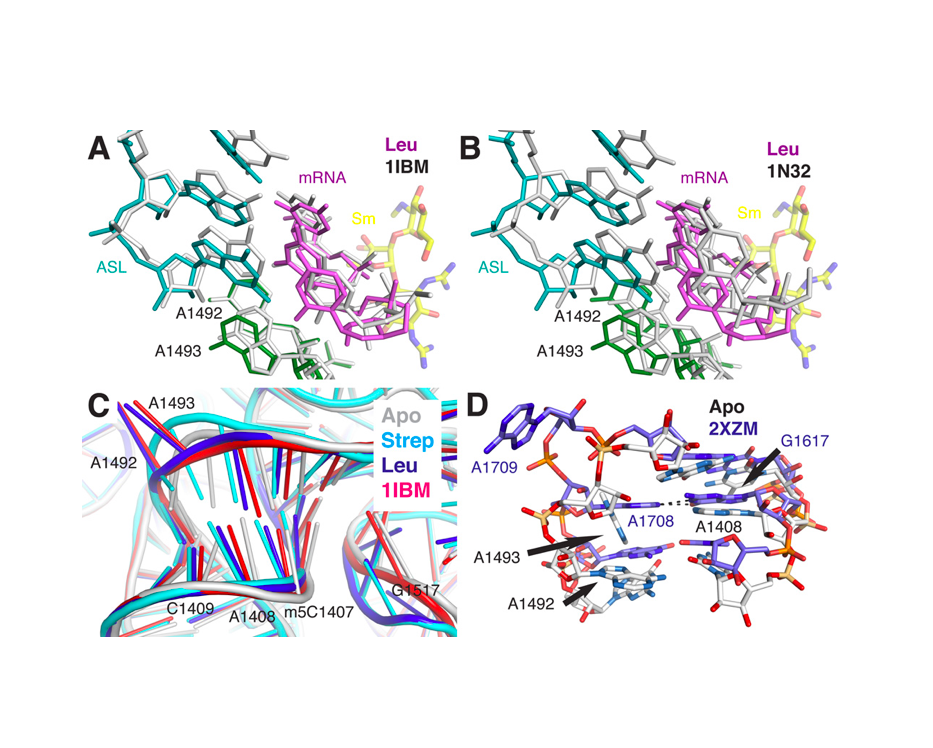
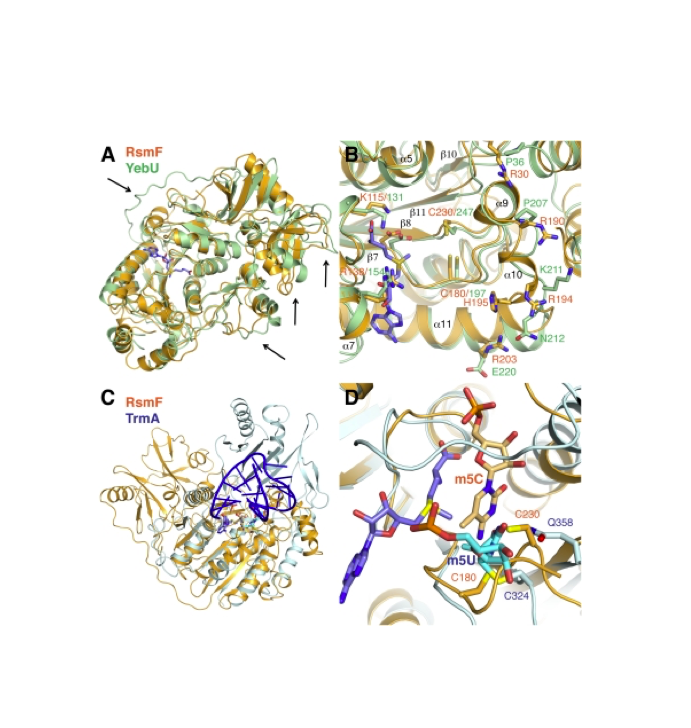
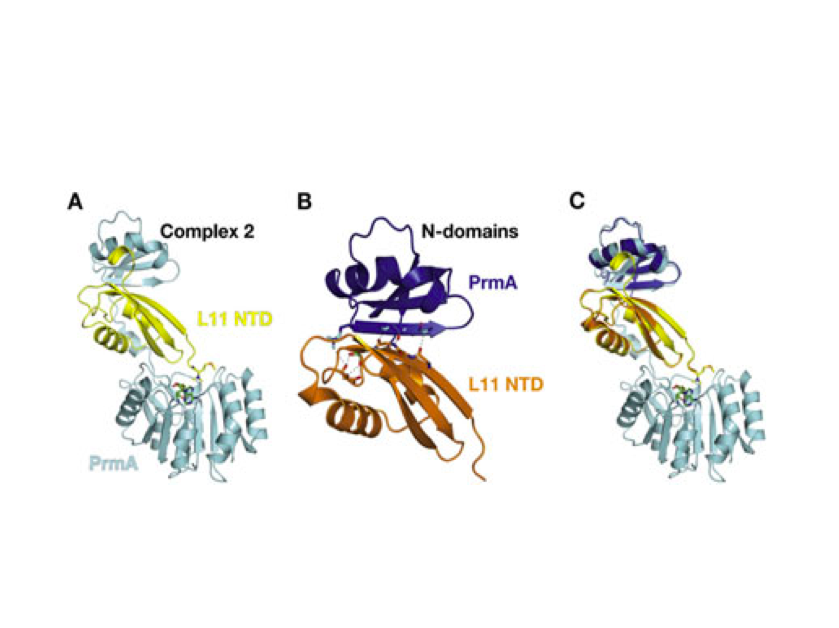
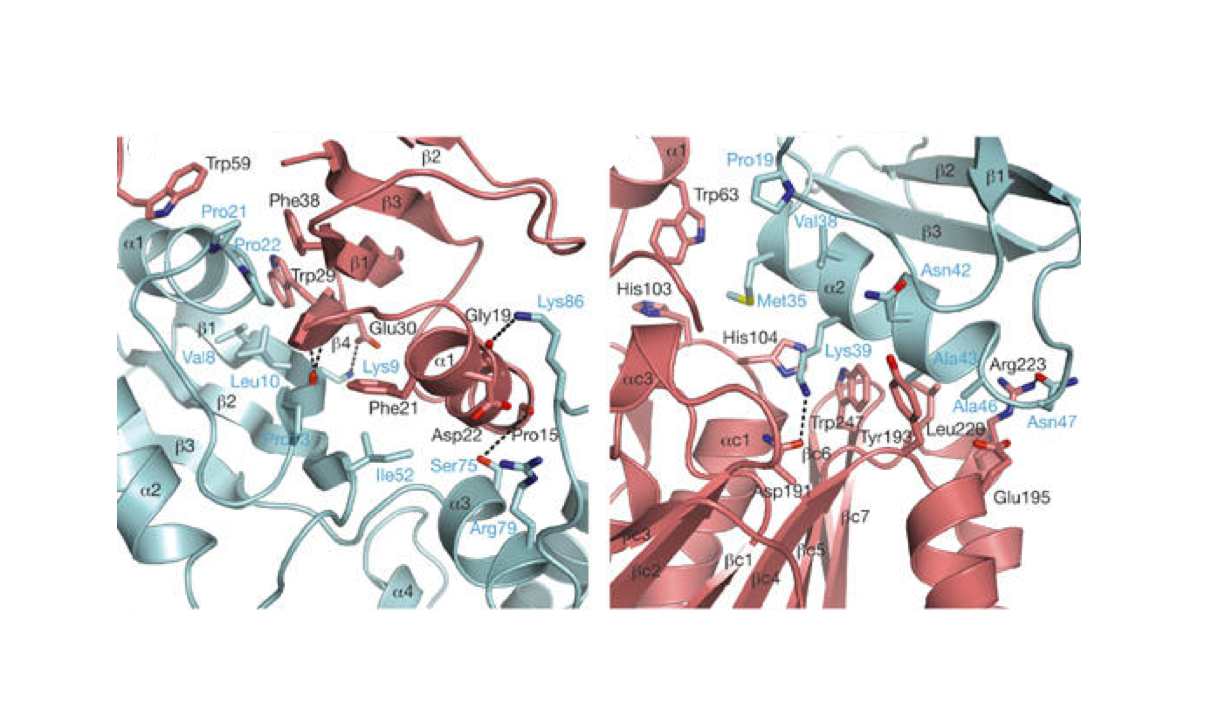
Nature Structural & Molecular Biology 2018
![]()
![]()
![]()
![]()
![]()
![]()
![]()
![]()
![]()
![]()
![]()
![]()
![]()
![]()
![]()
![]()
![]()
![]()
![]()
![]()
![]()
![]()
![]()
![]()
![]()